
Abstract
Alcohol ingestion activates the autonomic nervous system and the hypothalamic-pituitary-adrenal axis. This study examined naltrexone effects on alcohol-induced increases in physiological responses and their association with alcohol liking. Using a within-subjects design, heavy drinking men (N = 19) were maintained on each of three naltrexone doses (0, 50, and 100 mg, p.o.) over an 8-day inpatient stay. Within each naltrexone dose, subjects had three alcohol challenge sessions (none, moderate, high) in random order. Autonomic, subjective and endocrine measurements were collected regularly prior to and following alcohol administration. High-dose alcohol ingestion increased heart rate, diastolic blood pressure, skin temperature, ACTH, cortisol and liking of drink effects; responses following the moderate alcohol dose were less consistent. Naltrexone significantly dampened alcohol-induced increases in heart rate, diastolic blood pressure, hormone levels and subjective liking of drink effects. This dampening of cardiovascular and hormonal responses may contribute to the therapeutic effectiveness of naltrexone for reducing alcohol liking and decreasing relapse in alcohol-dependent persons.
Similar content being viewed by others

Main
The brain opioid system has been proposed to be part of the neurocircuitry involved in alcohol reward and heavy alcohol drinking (Froehlich and Wand 1997; Gianoulakis and deWaele 1994; Reid et al. 1991). Pharmacological studies indicate that opioid receptor antagonists, such as naloxone hydrochloride and naltrexone, decrease alcohol self-administration in animal models (Altshuler et al. 1980; Froehlich et al. 1990; Hubbell et al. 1986, 1991; Marfaing-Jallat et al. 1983; Samson and Doyle 1985; Weiss et al. 1990). Clinical trials demonstrated that naltrexone reduces alcohol drinking and self-reported “high,” alcohol craving, as well as relapse rates in recently abstinent outpatient alcohol-dependent patients (O'Malley et al. 1992; Volpicelli et al. 1992, 1995) . These findings led to naltrexone's approval by the Food and Drug Administration as a pharmacotherapeutic agent for the treatment of alcohol dependence. Naltrexone is thought to reduce alcohol-induced high and relapse by binding to opioid receptors in discrete brain regions associated with reward and craving, and to prevent receptor activation by alcohol-induced release of endogenous opioid peptides.
Alcohol induces a variety of acute physiological responses when consumed in moderate-to-high doses. It is well established that intoxicating doses of alcohol increase heart rate (Docter et al. 1966; Iwase et al. 1995; McCaul et al. 1989; Sellers et al. 1972; Turkkan et al. 1988; van de Borne et al. 1997). These heart rate increases are hypothesized to be positively associated with the reinforcing or appetitive motivational effects of alcohol (Peterson et al. 1996) . Alcohol has less predictable effects on blood pressure, with reports of blood pressure increases (Iwase et al. 1995; Sellers et al. 1972) and no effects following alcohol ingestion (Turkkan et al. 1988; van de Borne et al. 1997). Finally, alcohol-induced peripheral vasodilation and associated skin temperature increases have been consistently reported (Iwase et al. 1995; Turkkan et al. 1988) and is thought to be one explanation for the lack of a consistent rise in blood pressure after alcohol consumption.
Acute administration of intoxicating doses of alcohol also activates the hypothalamic-pituitary-adrenal (HPA) axis thereby increasing ACTH and glucocorticoid levels (Inder et al. 1995; Rivier et al. 1984; Waltman et al. 1993). It remains unclear whether alcohol-induced activation of the HPA axis has any relationship to its reinforcing properties. Rodent studies show that alcohol increases plasma ACTH levels and that exogenous administration of ACTH and fragments of ACTH enhances alcohol consumption (Maickel and Sprague 1995). Observations in humans show that the ethanol-induced rise in ACTH is associated with euphoria and enhanced alpha wave production (Lukas and Mendelson 1988). Moreover, glucocorticoids have been shown to have reinforcing properties, to increase alcohol consumption and to enhance mesolimbic dopamine generation (Deroche et al. 1995; Fahlke et al. 1994; Samson 1995). Thus it is plausible that alcohol-induced activation of the HPA axis also modulates the rewarding properties of alcohol.
In contrast to alcohol, opioid antagonists do not have significant effects on cardiovascular responses, including heart rate and blood pressure in primates (Byrd 1983) and humans (Coppola et al. 1994; Gritz et al. 1976; Resnick et al. 1974). However, acute oral or intravenous administration of opioid antagonists activates the HPA axis over several hours (King et al. 1998; Wand et al. 1998). At present, the effect of repeated opioid antagonist administration on basal HPA-axis activity is unknown. Moreover, it remains unknown how chronic opioid antagonist administration would alter alcohol-induced HPA activity.
The purpose of the present study was to characterize the effects of chronic naltrexone administration on alcohol-induced physiological activation, including heart rate, blood pressure, skin temperature, and the HPA axis. Additionally, since alcohol-induced activation of heart rate and the HPA-axis is hypothesized to be associated with the reinforcing effects of alcohol, naltrexone's effects on subjective reports of “liking” drink effects also are examined. Male heavy drinkers received chronic, 8-day administration of three naltrexone doses (0, 50, and 100 mg, p.o.) in randomized order, separated by a one-week wash-out period. Three separate alcohol challenge sessions (none, moderate and high alcohol dose) were conducted during each naltrexone dosing period.
METHODS
Subjects
Subjects were recruited through the media. Respondents (N = 99) who were aged 25–60 years old, reported moderate to heavy alcohol use (an average of five drinks per occasion or five drinking days per week), low levels of associated problems (MAST score < 5) (Selzer 1971) and no individuals with acute or chronic health problems were invited to participate in a clinical interview and medical examination to determine study eligibility. Exclusion criteria included: current use of any prescription medication, chronic health problems, liver enzyme levels over twice normal limits, pregnancy, or a major Axis I diagnosis on the Structured Clinical Interview for DSM-IIIR (Spitzer and Williams 1987), including alcohol or drug abuse/dependence. Seventy-two individuals were excluded from study participation due to a diagnosis of alcohol abuse/dependence and/or medical or psychiatric disqualification.
A total of 27 subjects qualified for study participation. Four subjects either dropped out or were dismissed from the research for reasons unrelated to study procedures. The present paper reports findings for male subjects only (n = 19). Females were not included in the data analyses because of the limited number of women enrolled in the study (N = 4) and gender differences in baseline neuroendocrine measures.
Table 1 summarizes participants’ demographic and substance use characteristics. Eight subjects were of minority ethnic/racial status. Subjects averaged 38.4 years of age and generally had a high school education or greater. Two-thirds were employed at the time of their research participation and almost all had either never been married or were separated or divorced. Subjects reported drinking an average of almost five standard drinks per drinking occasion, consumed alcohol on an average of 12 days per month, and had been drinking alcohol for an average of 20.4 years. A little more than half of the subjects smoked tobacco and averaged about three-quarters of a pack of cigarettes per day. Approximately one-third reported occasional marijuana use and 21% of the sample also used cocaine occasionally.
Subjects were informed that the study was intended to determine how drugs from different pharmacological classes modify alcohol's effects. All potential subjects signed an institution-approved informed consent document prior to any research participation and were paid for their time in the study. This research was conducted in accord with the ethical standards of the Johns Hopkins University School of Medicine Joint Committee on Clinical Investigations and with the Declaration of Helsinki.
Procedures
All study sessions were conducted at the General Clinical Research Center (GCRC) at the Johns Hopkins Bayview Medical Center. Subjects participated in the research over a six-week period. Each of three naltrexone doses was administered over an 8-day inpatient stay on the GCRC. There was a one-week outpatient washout period between each naltrexone dose period.
During each GCRC stay, admission was on Friday. The naltrexone dose scheduled for that week was administered daily at 8 PM for seven days starting on the day of admission. Over the course of the 8-day inpatient stay, subjects participated in alcohol challenge sessions on Monday, Wednesday, and Friday. Subjects were discharged from the GCRC on Saturday morning.
On each alcohol challenge session day, subjects were provided with a standard (no caffeine) breakfast at 7:30 AM and a snack at 10:00 AM. Neither cigarette smoking nor additional food or drink were permitted between 10:00 AM and 3:30 PM. Sessions began with the insertion of a heparinized catheter in a non-dominant forearm vein approximately 90 min prior to alcohol administration. Subjects were seated in a quiet room during testing. Alcohol administration began at 11:45 AM.
Drug Preparation and Administration
Each subject participated in three naltrexone dose conditions (0, 50, and 100 mg, p.o. orally) in random order. Each daily naltrexone dose was prepackaged, individually labeled, and dated using 50 mg naltrexone tablets and matching placebo (DuPont Pharma). Subjects ingested naltrexone under observation by the GCRC nursing staff at 8 PM, daily.
Subjects participated in three alcohol challenge sessions in random order at each naltrexone dose level. Each session's beverage was prepared by mixing the appropriate amount of pure ethanol (minus 3 ml which was added just prior to administration; see below) with juice to a volume of 16 ounces. The drink was separated into three equal amounts, and subjects had 5 min to drink each glass (McCaul et al. 1990). Time 0 and alcohol administration both refer to the end of this 15-min beverage ingestion period. To conceal the alcohol content of the drink, a wrist band soaked in ethanol was placed around the glass to deliver a strong alcohol odor and 1 ml of ethanol was floated on top of each glass to deliver an ethanol taste. Alcohol dose levels were 0.0, 0.6, and 1.2 g/kg for the first four subjects; dose levels were reduced to 0.0, 0.5, and 1.0 g/kg for the remaining participants due to excessive intoxication following the initial high alcohol dose level. For analyses, these dose conditions have been combined into no, moderate, and high alcohol doses. GCRC nurses, the study research assistant, and subjects were blind to all ethanol and naltrexone doses.
Dependent Measures
Subjective, psychomotor, physiological, and neuroendocrine measures were collected during each session. Psychomotor and subjective responses of male and female subjects have been reported previously (McCaul et al. 2000a).
Subjective Responses
The present paper reports findings of an analog rating of liking (“How much do you like the effects of the capsule/beverage you received?”) that demonstrated a naltrexone-alcohol interaction in earlier analyses of combined male and female subjects (McCaul et al. 2000a); current findings are based on male subjects only. Using a joystick, subjects were instructed to rate liking at the current time on a horizontal line anchored with “Not at all” on the left and “Extremely” on the right. Ratings were completed before and at 30-min intervals for three hours following beverage administration.
Autonomic Responses
Heart rate and skin temperature were obtained continuously from −60 to 180 minutes during each session and collapsed into 5-min intervals for data reduction. Blood pressure was obtained once every 5 min during this same time period. Baseline measures were recorded over a 30-min interval during session minutes −44 to −15. The drink period occurred over a 15-min interval during session minutes −14 to 0. Post-ingestion measures were obtained over a 3-hour period from session minutes +1 to +180; post-ingestion measures were summarized in 30-min intervals for data analyses. Heart rate and blood pressure were collected using automated measurement equipment (Sentry II; NBS Medical Inc., Costa Mesa, CA). Skin temperature was collected from a skin surface thermistor (YSI, Yellowsprings, OH) placed on the middle finger of the nondominant hand.
Hormone Levels
During each alcohol challenge session, subjects underwent periodic blood draws for subsequent hormone and alcohol analyses. Relative to completion of alcohol ingestion (time 0), blood samples for neuroendocrine and blood alcohol determinations were drawn at −30, −15, 0, 15, 30, 60, 90, 120, 150, and 180 min. Baseline ACTH and cortisol measurements represent the average of the values obtained at −30 and −15 min. Blood alcohol levels were determined at 30-min intervals following completion of the drink period.
Plasma concentrations of ACTH were assayed by a 2-site IRMA (Nichols immunoradiometric assay). Intra-assay and inter-assay coefficients of variance were less than 9%. Plasma concentrations of cortisol were measured by Radioimmunoassay (Diagnostic Products Corporation, Inc., Los Angeles, CA). Intra-assay and inter-assay coefficients of variation were 5.2% and 8.0%, respectively. Blood ethanol concentrations were measured by enzymatic determination (Sigma, St. Louis, MO).
Data Analysis
Statistical analyses was performed using STATA 5.0 and its Generalized Estimating Equations module. Analyses were based on general linear models for longitudinal measurement of data that take into consideration the correlation structure between measurements pertaining to the same individual over time. Because of baseline variability across subjects, heart rate, blood pressure, and skin temperature were analyzed as change scores from baseline.
No effect of the order of alcohol dose administration was observed in the current study; therefore, alcohol dose order is not included in the statistical model. The initial analytic model included alcohol dose, naltrexone dose, interactions between alcohol and naltrexone doses, and time. If the interaction terms were not significant, the final model was simplified to include only alcohol dose, naltrexone dose and time. Analyses examined the effects of each active alcohol and naltrexone dose compared to placebo effects. Significance was evaluated at p = .05.
RESULTS
Blood Alcohol Levels
Following moderate alcohol dose administration, blood alcohol level (BAL) increased to 0.048 mg/dL at 30 min following ingestion, and then declined gradually across the remainder of the session to 0.018 mg/dL. Mean blood alcohol level averaged across the moderate alcohol dose session was 0.030 mg/dL (S.D. = 0.023). Following high alcohol dose administration, BAL increased to 0.102 mg/dL at 60 min following ingestion, and remained elevated at 0.078 mg/dL following three hours. Mean blood alcohol level averaged across the high alcohol dose session was 0.076 mg/dL (S.D. = 0.049). There were no differences in overall-session mean or peak blood alcohol levels or time to peak BALs as a function of naltrexone dose condition (all p > .10).
Heart Rate
Effects of Alcohol on Heart Rate
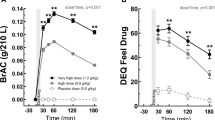
Figure 1 (panels A-C) show the heart rate during and following beverage ingestion as a function of alcohol and naltrexone dose levels. Following ingestion of placebo alcohol (Figure 1A), heart rate remained at baseline levels for the first hour and then decreased 6 bpm over the remaining two hours of the session. Moderate [Figure 1B, •] and high dose alcohol administration [Figure 1C, •] increased the mean heart rate by 5.2 bpm (S.E. = 0.82, Z = 8.77, p < .0001) and 5.3 bpm (S.E. = 0.83, Z = 8.73, p < .0001) averaged across the post-drinking period, when subjects were maintained on placebo naltrexone.

Effects of chronic naltrexone administration on heart rate and diastolic blood pressure following placebo (panels A and D), moderate dose (panels B and E), and high dose (panels C and F) alcohol ingestion. Symbols represent mean values averaged across subjects (N = 19) for each naltrexone dose condition at each time point. Naltrexone doses included placebo (•), 50 mg (○), and 100 mg (▾). Baseline measures were obtained over a 30-min interval during session minutes −44 to −15. The drink period (time 0) occurred over a 15-min interval during session minutes −14 to 0. Change scores were calculated for each of the nine naltrexone/alcohol conditions for each subject by subtracting the baseline measure for that session from post-ingestion measures.
Effects of Naltrexone on Heart Rate
Active naltrexone had no effect on baseline heart rate measured prior to alcohol administration or on mean heart rate following placebo alcohol ingestion. Active naltrexone dampened alcohol-induced increases in heart rate. There were significant interactions between the moderate alcohol dose and both active naltrexone doses; that is, naltrexone in interaction with the moderate alcohol dose produced a greater dampening effect than that predicted by either drug effect alone. In the moderate alcohol dose condition (Figure 1B), the mean heart rate following drink administration was decreased by 44% with 50 mg naltrexone (−3.14 bpm, S.E. = 1.15, Z = −2.72, p = .006) and by 30% with 100 mg naltrexone (−2.66 bpm, S.E. = 1.17, Z = −2.27, p = .023) relative to the mean heart rate in the placebo naltrexone condition.
In the high alcohol dose condition (Figure 1C), a similar trend was observed, with dampening of ethanol-induced heart rate increases of 37% with 50 mg naltrexone (−2.19 bpm, S.E. = 1.16, Z = −1.89, p = .058) and of 27% by 100 mg naltrexone (−2.00 bpm, S.E. = 1.19, Z = −1.68, p = .094) relative to placebo naltrexone. The dampening of alcohol-induced increases in heart rate was comparable in the two active naltrexone dose conditions (all p's > .10).
Blood Pressure
Effects of Alcohol on Blood Pressure
There was no effect of the moderate alcohol dose on mean diastolic blood pressure averaged across the post-ingestion period. The only significant effect of the moderate alcohol dose was a modest decrease in mean systolic blood pressure compared to the placebo alcohol session (−1.10 mm Hg, S.E. = 0.51, Z = −2.13, p = .03). In contrast, the high alcohol dose [Figure 1F, •] significantly increased mean diastolic blood pressure averaged over the 3 hour post-drink period (5.47 mm Hg, S.E. = 0.88, Z = 2.56, p = .01) relative to placebo alcohol administration [Figure 1D, •], when subjects were maintained on placebo naltrexone.
Effects of Naltrexone on Blood Pressure
Naltrexone had no effect on baseline systolic or diastolic blood pressure measured prior to alcohol administration. Naltrexone did not alter alcohol's effects on mean systolic blood pressure. However, similar to the effects observed on heart rate, there was an interaction of naltrexone and alcohol on mean diastolic blood pressure, resulting in significant dampening of ethanol-induced blood pressure increases. Following ingestion of the moderate alcohol dose (Figure 1E), mean diastolic blood pressure averaged across the session was decreased −2.31 mm Hg by 50 mg naltrexone (S.E. = 1.22, Z = −1.89, p = .058) and −4.09 mm Hg by 100 mg naltrexone (S.E. = 1.25, Z = −3.28, p = .001).
Following ingestion of the high alcohol dose (Figure 1F), mean diastolic blood pressure was decreased −2.65 mm Hg by 50 mg naltrexone (S.E. = 1.23, Z = −2.15, p = .031) and −3.39 mm Hg by 100 mg naltrexone (S.E. = 1.26, Z = −2.69, p = .007). This dampening effect on alcohol-induced increases in mean diastolic blood pressure was more pronounced following the high compared to the moderate naltrexone dose (p = .04).
Skin Temperature
Ingestion of the moderate and high alcohol doses increased mean skin temperature an average of 2.92°F (S.E. = 0.34, Z = 8.64, p< .0001) and 2.79°F (S.E. = 0.33, Z = 8.36, p < .0001) relative to mean skin temperature following placebo alcohol. There was no naltrexone effect on baseline skin temperature. Also, alcohol-induced skin temperature increases were not altered by naltrexone administration (data not shown).
Hormone Levels
Effects of Alcohol Ingestion on Hormone Levels
There were no significant effects of the moderate alcohol dose on mean plasma ACTH or cortisol levels (Figures 2B and 2E) compared to placebo alcohol. In contrast, following the high alcohol dose, plasma ACTH levels increased during the first 90 min post-ingestion and then declined to baseline values by 180 min, resulting in significant increases in mean ACTH levels (3.37 μg/dL; S.E. = 1.33, Z = 2.54, p = .01) (Figure 2C). Cortisol levels gradually increased over the entire 180-min period following the high alcohol dose; there were significant increases in mean cortisol levels (2.89 μg/dL; S.E. = 0.62, Z = 4.64, p < .0001) (Figure 2F) relative to placebo alcohol.

Effects of chronic naltrexone administration on plasma ACTH and cortisol following placebo (panels A and D), moderate dose (panels B and E), and high dose (panels C and F) alcohol ingestion. Symbols represent mean values averaged across subjects (N=19) in each naltrexone dose condition at each time point. Symbols to the left of time 0 represent baseline scores prior to beverage ingestion. Naltrexone doses include placebo (•), 50 mg (○), and 100 mg (▾).
Effects of Naltrexone on Hormone Levels
Naltrexone did not alter plasma ACTH or cortisol levels at baseline (16-hr after the last naltrexone dose and immediately prior to beverage ingestion), or following ingestion of placebo alcohol (Figures 2A and 2D). There were no main effects or interactions of naltrexone on mean ACTH responses following the moderate alcohol dose (Figure 2B); however, there was an interaction of naltrexone 50 mg and the high alcohol dose (−3.54 μg/dL; S.E. = 1.86, Z = −1.91, p = .057) (Figure 2C). In the high alcohol dose condition only, ACTH was significantly lower following naltrexone 50 mg compared to placebo naltrexone (−3.79 μg/dL; S.E. = 1.78, Z = −2.13, p = .03). There was no significant difference in ACTH response between naltrexone 50 mg and 100 mg.
There were no main effects or interactions of naltrexone on mean cortisol responses following the moderate alcohol dose (Figure 2E). Following ingestion of the high alcohol dose, there were significant interactions of naltrexone 50 mg and 100 mg on mean plasma cortisol responses averaged over the 3-hour session (Figure 2F). Specifically, in combination with the high dose of alcohol, mean cortisol levels were dampened by 89% with naltrexone 50 mg (−2.21 μg/dL; S.E. = 0.88, Z = −2.52, p = .012) and by 47% with naltrexone 100 mg (−2.10 μg/dL; S.E. = 0.88, Z = −2.40, p = .017) compared to placebo naltrexone. There was no significant difference in cortisol response between naltrexone 50 mg and 100 mg.
Subjective Measure of Alcohol Liking
Effects of Alcohol Ingestion on Liking
Mean ratings of liking the beverage/capsule effects increased following placebo alcohol administration (Figure 3A). Ratings of liking were significantly increased by the high alcohol dose (4.3; S.E. = 1.98, Z = 2.17, p = .03) (Figure 3C), but not the moderate dose relative to placebo alcohol (Figure 3B).

Effects of chronic naltrexone administration on subjects’ ratings of “like effects of beverage/capsule” following placebo (panel A), moderate dose (panel B), and high dose (panel C) alcohol ingestion. Symbols represent means averaged across subjects (N = 19) for each naltrexone dose condition at each time point. Symbols to the left of time 0 represent baseline scores prior to beverage ingestion. Naltrexone doses include placebo (•), 50 mg (○), and 100 mg (▾). Liking was scored on an analog scale from 0 (“not at all”) to 39 (“extremely”).
Effects of Naltrexone on Subjective Ratings
There was a significant main effect of 50 mg naltrexone on mean subjective reports of liking (−8.04; S.E. = 1.95, Z = −4.13, p < .0001), such that subjects reported reduced liking of the beverage/capsule effects across all alcohol dose conditions when maintained on 50 mg naltrexone compared with placebo naltrexone. A similar trend was observed across all alcohol dose conditions when subjects were maintained on 100 mg naltrexone (−3.62; S.E. = 1.98, Z = −1.83, p = .067) compared to placebo naltrexone. There was an interaction of the high alcohol dose and the high naltrexone dose, such that the decrement in mean liking in this dose combination was greater than that predicted by either drug alone (Figure 3C). This interaction was significant when naltrexone 100 mg was compared to placebo (−5.94; S.E. = 2.83, Z = −2.10, p = .036) or naltrexone 50 mg (−6.51; S.E. = 2.83, Z = −2.30, p = .021).
DISCUSSION
High dose alcohol ingestion significantly increased several autonomic and neuroendocrine measures in the present study; effects of the moderate alcohol dose were less consistent. Of importance, naltrexone blunted alcohol-induced increases in physiological and subjective responses. Specifically, heart rate, diastolic blood pressure, plasma ACTH, and cortisol increases following acute alcohol administration were significantly reduced when subjects were maintained on active compared to placebo naltrexone, although no clear dose response between 50 mg and 100 mg naltrexone emerged. The blunting of physiological effects by naltrexone was paralleled by a dampening of alcohol-induced increases in the subjective measure of alcohol liking. The relationship between this blunting of physiological effects and the therapeutic effectiveness of naltrexone for decreasing relapse in recently abstinent alcohol-dependent patients is intriguing.
In previous research, heart rate increases following alcohol administration have been associated with the rewarding effects of alcohol. For example, it is important that heart rate acceleration is greatest during the ascending limb of the BAL curve, the same time period that the stimulatory and rewarding properties of alcohol are most pronounced (Newlin and Thomson 1990). More generally, heart rate increases have been associated with appetitive responding for rewarding objects or events (Fowles 1980, 1988). These effects have been reported with monetary incentives (Fowles et al. 1982), responding for sucrose in neonates (Ashmead et al. 1980; Lipsitt et al. 1976), and the pharmacological effects of a variety of psychostimulant drugs (Pihl et al. 1995; Wise 1988).
One model for how this appetitive arousal model of heart rate increases is involved in alcohol reward involves the interaction of the opioid and dopamine neurotransmitter pathways. Specifically, acute alcohol administration increases beta-endorphin levels (Gianoulakis 1990), which in turn increase dopamine release (Spanagel et al. 1991). The dopamine system is believed to be central to the rewarding effects of a variety of commonly abused drugs (Koob and Bloom 1988; Wise 1996; Wise and Rompre 1989). Finally, this reward or motivational arousal associated with dopamine release stimulates heart rate increases similar to those observed with other appetitive events (i.e., money or sucrose delivery). Monetary incentives studied in Fowles et al. 1982 referenced above. “Appetitive” is used broadly to refer to objects/events organisms seek. By blocking opiate receptors in key brain regions involved in the dopamine reward pathway, naltrexone blocks this beta-endorphin-induced release of dopamine (Benjamin et al. 1993), in turn dampening the rewarding effects of alcohol and the associated heart rate increases.
There are several lines of research that support this appetitive arousal model of opioid involvement in alcohol reward. First, alcohol administration stimulates significantly greater heart rate increases in young adults at increased risk for alcoholism (Conrod et al. 1997; Finn and Pihl 1988; Peterson et al. 1996), and in alcohol-dependent subjects (Cohen et al. 1983; Kaplan et al. 1985; McCaul et al. 1989) than normal controls. Second, alcohol-induced beta-endorphin production is greater in alcohol-preferring than non-preferring animals (deWaele and Gianoulakis 1993; Gianoulakis et al. 1992) and in high-risk offspring of alcoholics compared to control offspring of nonalcoholics (Gianoulakis et al. 1996). In turn, alcohol-induced heart rate increases are highly correlated with alcohol-induced increases in beta-endorphin (Peterson et al. 1996). These findings suggest that persons at increased risk for alcohol dependence demonstrate more robust heart rate increases following alcohol administration and, by implication, have greater sensitivity to the rewarding effects of alcohol ingestion.
There also is growing evidence that stress and glucocorticoids are involved in reward through mesolimbic-dopamine chemistry. First, rodents self-administer glucocorticoids and develop place preference to this steroid class. Rodent studies have shown that glucocorticoids released during stress enhance mesolimbic dopamine generation (Samson 1995; Sorg and Kalivas 1991). This effect on mesolimbic dopamine release is reversed by adrenalectomy. It has been suggested that the ability of stress to increase mesolimbic dopamine production results in sensitization of the reward pathway to drugs of abuse (Deroche et al. 1992; Piazza et al. 1990, 1991a,b; Samson 1995; Shaham and Stewart 1994). Indeed, rats will drink more alcohol following stress and these post-stress alcohol-drinking levels are attenuated by adrenalectomy (Fahlke et al. 1994).
Second, young primates that develop high plasma cortisol levels in response to a stressful rearing environment subsequently demonstrate increased alcohol consumption as adults compared to primates with low plasma cortisol levels regardless of rearing condition (Fahlke et al. 2000).
Third, offspring of alcoholics have abnormal HPA axis dynamics, making higher cortisol levels in response to certain provocators including naloxone challenge (Wand et al. 1998), although these effects have not been consistently observed (Schuckit et al. 1987). Finally, alcoholics have an abnormal HPA axis, generating more cortisol during active drinking and acute withdrawal, but a blunted stress cortisol response following 3 – 4 weeks of alcohol abstinence (Lovallo et al. 2000).
In alcohol dependent persons, HPA-axis abnormalities experienced during early abstinence may contribute to dysphoria, craving and ultimately the high rate of relapse during this period (Marchesi et al. 1997; Tsigos and Chrousos 1995). It is intriguing that administration of a single dose of an opioid antagonist (Farren et al. 1999; King et al. 1998; Wand et al. 1998) results in acute stimulation of the HPA-axis, perhaps contributing to alleviation of craving as reported in earlier clinical trials (Volpicelli et al. 1992).
Our data demonstrated that, 16 hours following the most recent capsule ingestion, ACTH and cortisol levels are comparable following active naltrexone and placebo. This finding is congruent with observations of waning effects in HPA-axis stimulation within several hours of opioid antagonist administration. Despite the absence of a direct effect of naltrexone on the HPA-axis 16 hours following dosing, it is important that the antagonist is still able to block alcohol-induced activation of ACTH and cortisol.
What do these observations tell us about the mechanism of therapeutic action of naltrexone for the treatment of alcohol dependence? To date, most explanations for naltrexone's therapeutic effectiveness in alcohol dependent patients have centered on the direct role of the opioid system in alcohol reward. It is provocative to speculate that naltrexone's effect on the HPA-axis may also play a role in its therapeutic efficacy. In this model, craving is reduced following acute administration of naltrexone through its ability to transiently increase ACTH and cortisol levels. Simultaneously, naltrexone blocks ethanol's activation of ACTH and cortisol and in this manner may attenuate alcohol reward.
There are a number of design strengths and weaknesses in the present study. A major strength was the inpatient protocol that assured naltrexone dose compliance and minimized the risk of unauthorized alcohol or drug use. A second study strength was the within-subject design that crossed all alcohol and naltrexone doses within each subject, increasing the statistical power of our study to observe main and interaction effects of naltrexone and alcohol. A final strength was the chronic dosing model for naltrexone administration that approximates naltrexone's use in clinical populations. This procedure increased the likelihood that naltrexone and 6-β-naltrexol (the major metabolite of naltrexone) were at steady-state across alcohol challenge sessions, and decreased the risk of acute, naltrexone-induced nausea interfering with the study measures. Indeed, measurements obtained immediately prior to each alcohol challenge session confirmed that beta-naltrexol levels were equivalent across alcohol session days (McCaul et al. 2000b). Relatedly, the timing of alcohol challenge sessions 16 hours following the most recent naltrexone dose ingestion was chosen based on the half-life of beta-naltrexol (14 - 18 hours) and the clinical reality that most patients experience a considerable time lag between medication ingestion and onset of relapse drinking.
There also are three potential limitations to the current study. First, we studied heavy drinkers rather than alcohol-dependent subjects. At present, there is a lack of consensus on the most appropriate population for laboratory studies involving alcohol administration. Current Institutional Review Board and National Institute on Alcohol Abuse and Alcoholism guidelines discourage the conduct of alcohol challenge studies in alcohol-dependent persons. Further, there are significant interpretative problems, particularly involving autonomic and hormonal measurements, when alcohol-dependent laboratory subjects are undergoing alcohol withdrawal immediately prior to or during the protocol. The fact that results of the present study are similar to those of observed with alcohol-dependent patients provides face validity to our subject selection. Also, the growing clinical interest in prophylactic use of naltrexone for management of heavy drinking in nonalcohol-dependent persons increases the direct clinical relevance of the current findings (Bohn et al. 1994).
A second potential weakness of the current study is that approximately one-third of the subjects acknowledged lifetime marijuana use and 21% reported occasional cocaine use, although no subjects met abuse or dependence criteria for these drugs and no subject had positive urinalysis results during the study procedures. Thus, we can be reasonably confident that current study findings reflect the interaction of naltrexone and alcohol only and not the interaction with illicit drugs.
Finally, over half of the study subjects smoked tobacco. Several strategies were used to minimize the impact of tobacco use on study findings: smokers who used more than one pack per day were excluded from study participation; and smoking was prohibited for two hours prior to and throughout each alcohol challenge session. Any chronic perturbations of the autonomic or HPA measures would be comparable across alcohol dose conditions. Indeed, when smoking status was included in the analyses, no overall effect of smoking was observed.
In summary, the current findings support potential roles of several therapeutic mechanisms for naltrexone's effects on alcohol reward and relapse. In addition to naltrexone's direct impact on alcohol reward through blockade of alcohol-induced endogenous peptide activation of opioid receptors, it is plausible that naltrexone blockade indirectly dampens dopamine release and associated appetitive heart increases and alcohol-induced HPA activation. Both dopamine release and HPA activation have demonstrated reinforcing effects. Thus, naltrexone's effects on these alternative reward pathways may have important implications for long-term, sustained pharmacotherapeutic effects for the treatment of alcohol dependence.
References
Altshuler HL, Phillips PE, Feinhandler DA . (1980): Alteration of ethanol self-administration by naltrexone. Life Sci 26: 679–688
Ashmead DH, Reilly BM, Lipsitt LP . (1980): Neonates’ heart rate, sucking rhythm, and sucking amplitude as a function of the sweet taste. J Exp Child Psychol 29: 264–281
Benjamin D, Grant ER, Pohorecky A . (1993): Naltrexone reverses ethanol-induced dopamine release in the nucleus accumbens in awake freely moving rats. Brain Res 621: 137–140
Bohn MJ, Kranzler HR, Beazoglou D . (1994): Naltrexone and brief counseling to reduce heavy drinking. Am J Addict 3: 91–99
Byrd LD . (1983): Cardiovascular effects of naloxone, naltrexone and morphine in the squirrel monkey. Life Sci 32: 391–398
Cohen MJ, Schandler SL, Naliboff BD . (1983): Psychophysiological measures from intoxicated and detoxified alcoholics. J Stud Alcohol 44: 271–282
Conrod PJ, Peterson JB, Pihl RO, Mankowski S . (1997): Biphasic effects of alcohol on heart rate are influenced by alcoholic family history and rate of alcohol ingestion. Alcohol Clin Exp Res 21: 140–149
Coppola L, Cozzolino D, Guigliano D, Verrazzo G, Tirelli A, Giunta R, Buoninconti R, Torella R . (1994): Hemorheological and cardiovascular responses to beta-endorphin and naloxone in healthy subjects and in patients with essential hypertension. J Clin Endocrinol Metab 79: 826–830
Deroche V, Merinelli M, Maccari S . (1995): Stress-induced sensitization and glucocorticoids. J Neurosci 15: 181–188
Deroche V, Piazza PV, Maccari S, Le Moal M, Simon H . (1992): Repeated corticosterone administration sensitizes the locomotor response to amphetamine. Brain Res 584: 309–313
deWaele JP, Gianoulakis C . (1993): Effects of single and repeated exposures to ethanol on the hypothalamic beta-endorphin and CRH release by the C57BL/6 and DBA/2 strains of mice. Neuroendocrinology 57: 700–709
Docter RF, Naitoh P, Smith JC . (1966): Electroencephalographic changes and vigilance behavior during experimentally induced intoxication with alcoholic subjects. Psychosom Med 28: 605–615
Fahlke C, Engel JA, Eriksson CJP, Hard E, Soderpalm B . (1994): Involvement of corticosterone in the modulation of ethanol consumption in the rat. Alcohol 11: 195–202
Fahlke C, Lorenz JG, Long J, Champoux M, Suomi SJ, Higley JD . (2000): Rearing experiences and stress-induced plasma coritsol as early risk factors for excessive alcohol consumption in nonhuman primates. Alcohol Clin Exp Res 24: 644–650
Farren CK, O'Malley SS, Grebski G, Maniar S, Porter M, Kreek MJ . (1999): Variable dose naltrexone-induced hyupothalamic-pituitary-adrenal stimulation in abstinent alcoholics: A preliminary study. Alcohol Clin Exp Res 23: 502–508
Finn PR, Pihl RO . (1988): Risk for alcoholism: A comparison between two different groups of sons of alcoholics on cardiovascular reactivity and sensitivity to alcohol. Alcohol Clin Exp Res 12: 742–747
Fowles DC . (1980): The three arousal models: Implications for Gray's two-factor learning theory for heart rate, electrodermal activity, and psychopathy. Psychophysiology 17: 87–104
Fowles DC . (1988): Psychophysiology and psychopathology: A motivational approach. Psychophysiology 25: 373–391
Fowles DC, Fisher AE, Tranel DT . (1982): The heart beats to reward: The effect of monetary incentive on heart rate. Psychopharmacology 19: 506–513
Froehlich JC, Harts J, Lumeng L, Li TK . (1990): Naloxone attenuates voluntary ethanol intake in rats selectively bred for high ethanol preference. Pharmacol Biochem Behav 35: 385–390
Froehlich JC, Wand GS . (1997): The neurobiology of ethanol-opioid interactions in ethanol reinforcement. Alcohol Clin Exp Res 20: 181A–186A
Gianoulakis C . (1990): Characterization of the effects of acute ethanol administration on the release of beta-endorphin peptides by the rat hypothalamus. Eur J Pharmacol 180: 21–29
Gianoulakis C, deWaele J . (1994): Genetics of alcoholism. Metab Brain Dis 9: 105–131
Gianoulakis C, deWaele JP, Kiianmaa K . (1992): Differences in the brain and pituitary beta-endorphin system between the alcohol-preferring AA and alcohol-avoiding ANA rats. Alcohol Clin Exp Res 16: 453–459
Gianoulakis C, Krishnan B, Thavundayil J . (1996): Enhanced sensitivity of pituitary beta-endorphin to ethanol in subjects at high risk of alcoholism. Arch Gen Psychiatry 53: 250–257
Gritz ER, Shiffman SM, Jarvik ME, Schlesinger J, Charuvastra VC . (1976): Naltrexone: Physiological and psychological effects of single doses. Clin Pharmacol Therap 19: 773–776
Hubbell CL, Czirr SA, Hunter GA, Beaman CM, LeCann NC, Reid LD . (1986): Consumption of ethanol solution is potentiated by morphine and attenuated by naloxone persistently across repeated daily administrations. Alcohol 3: 39–54
Hubbell CL, Marglin SH, Spitalnic SJ, Abelson ML, Wild KD, Reid LD . (1991): Opioidergic, serotonergic, and dopaminergic manipulations and rats’ intake of a sweetened alcoholic beverage. Alcohol 8: 355–367
Inder WJ, Joyce PR, Ellis MJ, Evans MJ, Livesey JH, Donald RA . (1995): The effects of alcoholism on the hypothalamic-pituitary-adrenal axis: Interactions with endogenous opioid peptides. Clin Endocrinol 43: 283–290
Iwase S, Matsukawa T, Ishihara S, Tanaka A, Tanabe K, Danbara A, Matsuo M, Sugiyama Y, Mano T . (1995): Effect of oral ethanol intake on muscle sympathetic nerve activity and cardiovascular functions in humans. J Auton Nerv Syst 54: 206–214
Kaplan HL, Sellers EM, Hamilton C, Naranjo CA, Dorian P . (1985): Is there acute tolerance to alcohol at steady state? J Stud Alcohol 46: 253–256
King CA, Schleuger J, Disla I, Kreek MJ . (1998): Acute neuroendocrine effects of oral naltrexone in family history positive and negative social drinkers. Alcohol Clin Exp Res 22: 120A
Koob GF, Bloom FE . (1988): Cellular and molecular mechanisms of drug dependence. Science 242: 715–723
Lipsitt LP, Reilly BM, Butcher MJ, Greenwood MM . (1976): The stability and interrelationships of newborn sucking and heart rate. Dev Psychobiol 9: 305–310
Lovallo WR, Dickensheets SL, Myers DA, Thomas TL, Nixon SJ . (2000): Blunted stress cortisol response in abstinent alcoholic and plysubstance-abusing men. Alcohol Clin Exp Res 24: 651–658
Lukas SE, Mendelson JH . (1988): Electroencephalographic activity and plasma ACTH during ethanol-induced euphoria. Biol Psychiatry 23: 141–148
Maickel RP, Sprague JE . (1995): Role of ACTH fragments in alcohol consumption. In Hunt WA, Zakari S (eds), Stress, Gender and Alcohol-seeking Behavior, vol. 29. Washington, DC, Government Printing Office, pp 167–180
Marchesi C, Chiodera P, Ampollini P, Volpi R, Coiro V . (1997): Beta-endorphin, adrenocorticotropic hormone and cortisol secretion in abstinent alcoholics. Psychiatry Res 72: 187–194
Marfaing-Jallat P, Miceli D, LeMagnen J . (1983): Decrease in ethanol consumption by naloxone in naive and dependent rats. Pharmacol Biochem Behav 18: 537–539
McCaul ME, Turkkan JS, Stitzer ML . (1989): Conditioned opponent responses: Effects of placebo challenge in alcoholic subjects. Alcohol Clin Exp Res 13: 631–635
McCaul ME, Turkkan JS, Svikis DS, Bigelow GE . (1990): Alcohol and secobarbital effects as a function of familial alcoholism: Acute psychophysiological effects. Alcohol Clin Exp Res 14: 704–712
McCaul ME, Wand GS, Eissenberg T, Rohde CA, Cheskin LJ . (2000a): Naltrexone alters subjective and psychomotor responses to alcohol in heavy drinking subjects. Neuropsychopharmacology 22: 480–492
McCaul ME, Wand GS, Rohde C, Lee SM . (2000b): Serum 6-beta-naltrexol levels are related to alcohol responses in heavy drinkers. Alcohol Clin Exp Res 24: 1385–1391
Newlin DB, Thomson JB . (1990): Alcohol challenge with sons of alcoholics: A critical review and analysis. Psychol Bull 108: 383–402
O'Malley SS, Jaffe AJ, Chang G, Schottenfeld RS, Meyer RE, Rounsaville B . (1992): Naltrexone and coping skills therapy for alcohol dependence. A controlled study. Arch Gen Psychiatry 49: 881–887
Peterson JB, Pihl RO, Gianoulakis C, Conrod P, Finn PR, Stewart SH, LeMarquand DG, Bruce KR . (1996): Ethanol-induced change in cardiac and endogenous opiate function and risk for alcoholism. Alcohol Clin Exp Res 20: 1542–1552
Piazza PV, Deminiere J-M, Le Moal M, Simon H . (1991a): Factors that predict individual vulnerability to amphetamine self-administration. Science 245: 1511–1533
Piazza PV, Deminiere J-M, Le Moal M, Simon H . (1991b): Stress- and pharmacologically-induced behavioral sensitization increases vulnerability to acquisition of amphetamine self-administration. Brain Res 514: 22–26
Piazza PV, Deminiere J-M, Maccari S, Le Moal M, Simon H . (1990): Individual reactivity to novelty predicts probability of amphetamine self-administration. Behav Pharmacol 1: 339–345
Pihl RO, Giancola P, Peterson JB . (1995): Cardiovascular reactivity as a predictor of alcohol consumption in a taste test situation. J Clin Psychol 50: 280–286
Reid LD, Delconte JD, Nichols ML, Bilsky EJ, Hubbell CL . (1991): Tests of opioid deficiency hypotheses of alcoholism. Alcohol 8: 247–257
Resnick R, Volavka J, Freedman AM, Thomas M . (1974): Studies of EN-1639A (Naltrexone): A new narcotic antagonist. Am J Psychiatry 131: 646–650
Rivier C, Bruhn T, Vale W . (1984): Effect of ethanol on the hypothalamic-pituitary-adrenal axis in the rat: Role of corticotropin-releasing factor (CRF). J Pharmacol Exp Ther 229: 127
Samson H . (1995): Environmental effects on drinking patterns: Stress and sensitization. In Hunt WA, Zakari S (eds), Stress, Gender and Alcohol-seeking Behavior, vol. 29. Washington, DC, Government Printing Office, pp 277–292
Samson HH, Doyle TF . (1985): Oral ethanol self-administration in the rate: Effect of naloxone. Pharmacol Biochem Behav 22: 91–100
Schuckit MA, Gold E, Risch C . (1987): Plasma cortisol levels following ethanol in sons of alcoholics and controls. Arch Gen Psychiatry 44: 942–945
Sellers EM, Carr GC, Bernstein JG, Seller S, Koch-Weser J . (1972): Interaction of chloral hydrate and ethanol in man. Clin Pharmacol Ther 13: 50–58
Selzer ML . (1971): The Michigan Alcoholism Screening Test: The quest for a new diagnostic instrument. Am J Psychiatry 127: 1653–1658
Shaham Y, Stewart J . (1994): Exposure to mild stress enhances the reinforcing efficacy of intravenous heroin self-administration in rats. Psychopharmacology (Berl) 114: 523–527
Sorg BA, Kalivas PW . (1991): Effects of cocaine and footshock stress on extracellular dopamine levels in ventral striatum. Brain Res 559: 29–36
Spanagel R, Herz A, Bals-Kubik R, Shippenberg TS . (1991): Beta-endorphin-induced locomotor stimulation and reinforcement are associated with an increase in dopamine release in the nucleus accumbens. Psychopharmacology 104: 51–56
Spitzer RL, Williams JBW . (1987): Structured Interview for DSM-IIIR (SCID). New York, New York State Psychiatric Institute, Biometrics Research
Tsigos C, Chrousos GP . (1995): The neuroendocrinology of the stress response. In Hunt WA, Zakari S (eds), Stress, Gender and Alcohol-seeking Behavior, vol. 29. Washington, DC, Government Printing Office, pp 103–124
Turkkan JS, Stitzer ML, McCaul ME . (1988): Psychophysiological effects of oral ethanol in alcoholics and social drinkers. Alcohol Clin Exp Res 12: 32–38
van de Borne P, Mark AL, Montano N, Mion D, Somers VK . (1997): Effects of alcohol on sympathetic activity, hemodynamics, and chemoreflex sensitivity. Hypertension 29: 1278–1283
Volpicelli JR, Alterman AI, Hayashida M, O'Brien CP . (1992): Naltrexone in the treatment of alcohol dependence. Arch Gen Psychiatry 49: 876–880
Volpicelli JR, Watson NT, King A, Sherman CE, O'Brien CP . (1995): Effect of naltrexone on alcohol “high” in alcoholics. Am J Psychiatry 152: 613–615
Waltman C, Blevin L, Boyd G, Wand GS . (1993): The effects of mild ethanol intoxication on the Hypothalamic-Pituitary-Adrenal Axis in nonalcoholic men. J Clin Endocrinol Metab 77: 518–522
Wand GS, Mangold D, ElDeiry S, McCaul ME, Hoover D . (1998): Family history of alcoholism and hypothalamic opioidergic activity. Arch Gen Psychiatry 55: 1114–1119
Weiss F, Mitchiner M, Bloom FE, Koob GF . (1990): Free-choice responding for ethanol versus water in alcohol preferring (P) and unselected Wistar rats is differentially modified by naloxone, bromocriptine, and methysergide. Psychopharmacology 101: 178–186
Wise RA . (1988): Psychomotor stimulant properties of addictive drugs. Ann N Y Acad Sci 537: 228–234
Wise RA . (1996): Neurobiology of addiction. Curr Opin Neurobiol 6: 243–251
Wise RA, Rompre PP . (1989): Brain dopamine and reward. Annu Rev Psychol 40: 191–225
Acknowledgements
This work was conducted at The Johns Hopkins Bayview Medical Center GCRC, supported by MO1 RR02719. Additional support was provided by NIAAA R01-AA09569 (MEM), NIAAA R01-11855 (MEM), NIAAA R01-AA10158 (GSW), The Alcoholic Beverage Medical Research Foundation (GSW), and a generous gift from the Kenneth Lattman Foundation (GSW). Naltrexone 50 mg tablets were supplied at no cost by DuPont Pharma. The authors thank Thomas Eissenberg, Ph.D., Lawrence Cheskin, M.D., Joseph Bezold, and John Yingling for their help in conducting this research.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
McCaul, M., Wand, G., Stauffer, R. et al. Naltrexone Dampens Ethanol-Induced Cardiovascular and Hypothalamic-Pituitary-Adrenal Axis Activation. Neuropsychopharmacol 25, 537–547 (2001). https://doi.org/10.1016/S0893-133X(01)00241-X
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1016/S0893-133X(01)00241-X
Keywords
This article is cited by
-
New steps for treating alcohol use disorder
Psychopharmacology (2018)
-
Chronic alcohol consumption from adolescence-to-adulthood in mice - hypothalamic gene expression changes in the dilated cardiomyopathy signaling pathway
BMC Neuroscience (2014)
-
Pharmacogenetics of Naltrexone in Asian Americans: A Randomized Placebo-Controlled Laboratory Study
Neuropsychopharmacology (2012)
-
Pharmacological effects of naltrexone and intravenous alcohol on craving for cigarettes among light smokers: a pilot study
Psychopharmacology (2007)
-
Moderate doses of ethanol fail to increase plasma levels of neurosteroid 3α-hydroxy-5α-pregnan-20-one-like immunoreactivity in healthy men and women
Psychopharmacology (2006)
