
Abstract
Semiconductor devices continue to press into the nanoscale regime, and new applications have been proposed for which a single dopant atom acts as the functional part of the device1,2,3. Moreover, because shallow donors and acceptors are analogous to hydrogen atoms, experiments on small numbers of dopants have the potential to be a testing ground for fundamental questions of atomic and molecular physics4,5. Although dopant properties are well understood with respect to the bulk, the study of configurations of dopants in small numbers is an emerging field6,7. Here we present local capacitance measurements of electrons entering silicon donors in a gallium arsenide heterostructure. To the best of our knowledge, this study is the first example of single-electron capacitance spectroscopy carried out directly with a scanning probe tip8. The precise position with respect to tip voltage of the observed single-electron peaks varies with the location of the probe, reflecting a random distribution of silicon within the donor plane. In addition, three broad capacitance peaks are observed independent of the probe location, indicating clusters of electrons entering the system at approximately the same voltages. These broad peaks are consistent with the addition energy spectrum of donor molecules, effectively formed by nearest-neighbour pairs of silicon donors.
Similar content being viewed by others
Main
Electron tunnelling spectroscopy through isolated dopants has been observed in transport studies9,10. In addition, Geim and co-workers identified resonances due to two closely spaced donors, effectively forming donor molecules11. Rather than measuring transport current, our scanning-probe method is essentially a capacitance technique8,12. In contrast to the work of Geim et al., the measurements show discernible peaks attributed to successive electrons entering the molecules.
Our method is an extension of scanning charge accumulation imaging12. Figure 1a shows the experiment schematically. The key component is a metallic tip with an apex of radius∼50 nm, connected directly to a charge sensor, which achieves a sensitivity of  (ref. 13). For the capacitance spectroscopy measurements reported here, the tip’s position is fixed (that is, not scanned) at a distance of ∼1 nm from the sample surface. We then monitor the tip’s a.c. charge qtip in response to an a.c. excitation voltage Vexc applied to an underlying electrode, as a function of d.c. bias voltage Vtip. As detailed in the Methods section, if the quantum system below the tip can accommodate additional charge, the excitation voltage causes it to resonate between the system and the underlying electrode—giving rise to an enhanced capacitance, C≡qtip/Vexc.
(ref. 13). For the capacitance spectroscopy measurements reported here, the tip’s position is fixed (that is, not scanned) at a distance of ∼1 nm from the sample surface. We then monitor the tip’s a.c. charge qtip in response to an a.c. excitation voltage Vexc applied to an underlying electrode, as a function of d.c. bias voltage Vtip. As detailed in the Methods section, if the quantum system below the tip can accommodate additional charge, the excitation voltage causes it to resonate between the system and the underlying electrode—giving rise to an enhanced capacitance, C≡qtip/Vexc.

a, A schematic diagram of the key layers in the gallium arsenide [001] heterostructure sample and the measurement technique. An excitation voltage can cause charge to resonate between the Si donor layer and a two-dimensional (2D) layer, which represents an ideal base electrode. This results in image charge appearing on a sharp conducting PtIr tip. A cryogenic transistor attached directly to the tip is used to measure the charging. The donor layer consists of silicon atoms confined to a plane with respect to the z direction, but randomly positioned with respect to the x–y direction with an average density of 1.25×1016 m−2. At zero applied voltage, at least 90% of the Si atoms are ionized (that is, have donated an electron), as discussed in Supplementary Information, Fig. S1. Magnetocapacitance measurements conducted in the kilohertz frequency range indicate negligible donor-layer conductivity for identical samples cut from the same wafer. b, A more detailed conduction-band diagram of the sample. The excitation voltage is applied to a degenerately doped substrate that acts as a metallic electrode. Above this is the 2D electron layer. It is separated from the metallic substrate by a superlattice tunnelling barrier; the tunnelling rate into the 2D layer is an order of magnitude greater than the 20 kHz excitation frequency we used. Hence for this experiment, the 2D layer can be regarded as being in ohmic contact with the substrate. c, A schematic diagram of the area probed by the technique, with Si donors represented as hydrogenic potentials. For our experimental geometry, the radius of the area over which we are probing is set mostly by the tip–donor-layer separation, which is approximately 60 nm (refs 25, 26). Within this area, on average we expect 140 donors.
The first observations of donor-layer capacitance peaks acquired with our method probed a gallium arsenide heterostructure sample grown by molecular beam epitaxy, for which the donor layer was bulk-doped with Si at a density of 1024 m−3. To achieve better characterized results that would be more conducive to analysis, we used a GaAs [001] heterostructure sample that contains a high-mobility 2D electron layer, which serves as an ideal base electrode for the measurement. The donor plane consists of delta-doped Si of areal density 1.25×1016 m−2 within an Al0.3Ga0.7As layer. Samples from this wafer were used for all measurements reported here, with the sample and tip immersed in liquid helium-3 at a temperature of 290 mK. The conduction band profile is shown in Fig. 1b. Figure 1c shows an example of the energy landscape of the Si donor layer.
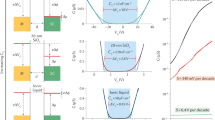
Figure 2a shows a representative capacitance curve; the measurements consistently showed three broad peaks in the vicinity of 0.5–0.9 V, labelled A, B and C. Supplementary Information, Fig. S1 shows the behaviour over a larger voltage range and details of the phase of the signal are given in Supplementary Information, Fig. S2. To help explain the physical origin of the broad peaks, we first examine the fine-structure peaks, as shown in Fig. 2b. The two curves in the figure were acquired under identical conditions but with a time delay of nine hours. We see that most, but not all, of the peaks are reproduced. Figure 2c shows three curves acquired at the voltage marked by the red arrow in Fig. 2b, with the average shown to the right. The data are consistent in magnitude and peak shape with the resonance expected for single-electron tunnelling14. Supplementary Information, Fig. S3a shows more details of the comparison between fine-structure peaks and the single-electron curve. Figure 2d is similar to Fig. 2a, except that here we show the average capacitance curve obtained by averaging measurements acquired at three different locations. For comparison, the curve is superposed with a capacitance curve acquired with a micrometre-size gate in place of the tip; we see that the gated data show only a step feature.

a, Representative local capacitance curve measured at a single tip position, with an excitation voltage amplitude of Vexc=15 mV r.m.s. The local measurements consistently showed three broad peaks labelled A, B and C. b, Capacitance curves acquired at the same position as in a, but over the indicated expanded voltage range. To investigate the structure in detail, here we used a smaller excitation amplitude of 3.8 mV r.m.s. Two curves are shown, which were acquired under identical conditions, but with a time delay of nine hours. Much of the fine structure is reproduced, with asterisks marking missing or shifted peaks. These changes probably reflect long-timescale charging and discharging of DX centres18. c, Three curves acquired near the voltage marked by the red arrow in b (same location) with an excitation voltage of 3.8 mV. The vertical scale has been converted to charge units, qtip. The plot to the right shows the average of the three measured curves, compared with a model curve which shows the semi-elliptical peak shape expected for single-electron tunnelling14; see Supplementary Information, Fig. S3a for a more detailed description. d, Capacitance measured with our local probe superposed with a curve acquired with a micrometre-size gate in place of the tip. To show clearly the characteristic structure of the broad peaks, here the probe measurement is the average of data acquired at three different locations. The averaging reduces the amplitude of the fine-structure peaks, which shift in voltage in a random way at different locations. For both curves, the excitation voltage amplitude was Vexc=15 mV r.m.s.; the voltage scales are plotted relative to the effective zero voltage, compensating for the contact potentials between the materials. The curves have different voltage ranges and the vertical scale of the probe measurement is exaggerated greatly relative to the gated measurement, consistent with differences in the probed area and with distinct lever-arm parameters for the two measurements (see the Methods section). The gated measurement effectively provides a background curve, as discussed in Supplementary Information, Figs S1 and S2.
Given that the fine-structure peaks are consistent with individual electrons entering the donor layer, a natural explanation for the broader peaks, A, B, and C, is that they are formed by clusters of several electrons entering at nearly the same energy. If we convert capacitance to charge units, we find that peaks A and B each correspond to about 15 electrons entering the donor system; peak C is larger. What physics could gives rise to the broad resonances? It is possible that dense groupings of the donors result in electron puddles, acting as small quantum dots15. In this scenario, an ensemble of puddles with similar addition energy spectra could explain the peaks. However, given that the positions of the donors are random, as shown schematically in Fig. 1c, it seems unlikely that 15 such puddles would form within a radius of only 60 nm with sufficiently similar characteristics. Considering the opposite limit, a candidate for identical quantum objects is single silicon donors. However, according to Lieb’s theorem, the maximum negative ionization for a molecule with K ions is Z+K−1, where Z is the total nuclear charge of the ions5. For a single donor this gives unity, corresponding to the H− system. That would give only two peaks in a capacitance measurement, which is inconsistent with our observations (neglecting for the moment the perturbation of the tip). However, if we consider closely spaced Si donors effectively forming two-donor molecules (2DMs), then K=2 and Z=2, giving Z+K−1=3, which would comfortably allow up to five peaks.
To develop a model for a theoretical addition spectrum of 2DMs, our approach is to use the configuration-interaction method in the context of the effective mass theory4,16,17,18. In this approximation, each donor is regarded as a hydrogenic atom with an effective Bohr radius a0*=4πɛ0κℏ2/m*e2 and effective Rydberg energy Ry*=e2/8πɛ0κ a0*, where ɛ0 is the permittivity of free space, e is the electron charge, m* is the electron effective mass and κ is the dielectric constant. In our sample, Si donors reside in Al0.3Ga0.7As, for which a0*=7.3 nm and Ry*=8.1 meV (ref. 19). This energy scale is much greater than the thermal energy at our experimental temperature.
Before introducing the full model, we consider the likelihood of finding donors in our sample spaced at a distance equal to or less than an effective Bohr radius. For simplicity, here we assume that the donors are confined to a perfect plane with respect to the z direction and distributed randomly with respect to x and y. In reality, the donors have a distribution in the z direction of roughly a0*; however, the simplification does not have a large impact on our analysis, as discussed in the Methods section. The planar density of 1.25×1016 m−2 implies an average spacing of 8.9 nm, which is comparable to a0*. So we expect many donors will have a nearest neighbour sufficiently close to form a 2DM. This fraction must be balanced against the fraction of donors that will have more than one nearby neighbour, as these will have qualitatively different addition spectra. Supplementary Information, Fig. S3b shows the statistical distributions of nearest-neighbour distances. By integrating over the appropriate distribution, we find that 38% of the donors are expected to have zero or one nearest neighbour within a0*. This is the relevant percentage for our 2DM model. Considering that the tip detects the charging of approximately 140 donors within the probed area, we expect on average to probe 0.38×140/2≈26 2DMs. Similarly, we expect on average to probe 12 three-donor molecules (3DMs), six four-donor molecules and still smaller numbers of larger clusters.
Figure 3a shows the configuration-interaction calculations of the 2DM addition energies for all bound electrons as a function of separation d. The calculations include an image charge to approximate the potential applied by the tip, which tends to increase the electron confinement within the molecule. We see that each molecule can accommodate four electrons. This is not surprising given that an isolated H atom, and a single Si donor, can accommodate two electrons. However, over the range of separations shown in Fig. 3a, without including the approximate tip potential, the calculations predict that each 2DM would hold only two electrons (see Supplementary Information, Fig. S3c). Supplementary Information, Fig. S3d shows more details of the tip confinement potential.

a, Configuration-interaction calculations of the 2DM addition energies for all bound electrons as a function of separation of the two ions d. The calculations include an image charge to approximate the potential applied by the tip (right inset). The model predicts four bound electrons for each donor molecule, as indicated by the schematic diagram (bottom). Including the approximate tip potential is key, as without the extra confinement the calculations show only two bound electrons, as shown in Supplementary Information, Fig. S3c. b, Schematic representation of the full modelling procedure. We start from a random ensemble of donors and group them into nearest-neighbour pairs to form molecules k. The addition energies shown in a are used to assign the isolated addition energy of each molecule, ɛ1k, ɛ2k and so on. Last, the model includes the Coulomb energy shift from all non-nearest neighbours; we account for the fact that this shift will be different for successive electrons owing to changes in the screening charge of non-nearest-neighbour donors, as described in the Methods section. c, Comparison between representative measured data acquired at a single tip position (black) and representative modelled data (red); the modelled curves were calculated based on two different random configurations of donors, labelled config. (i) and config. (ii). To enable a direct comparison, we subtract the background capacitance from the measurements and plot the voltage in units of effective Rydbergs (scale factor αtip/8.1×10−3 V/Ry*). The effective voltage increment for both the measured and modelled curves is approximately 0.2 Ry*. We see that the model predicts distinct broad peaks corresponding to the average electron addition spectra of the 2DMs; these peaks are reasonably qualitatively consistent with the observed peaks A, B and C. In contrast, the smaller fine-structure peaks, which correspond to individual charges entering the system, are different for different random configurations of the donors. d, Comparison between measured and modelled data averaged over multiple locations/configurations. The measured curve is the average of three locations (the same data as shown in Fig. 2d). The model curve is the average of hundreds of random ensembles, as described in the Methods section. Similarly to part c, we have subtracted the background capacitance from the measurements and plotted the voltage in units of effective Rydbergs. Moreover, here we show both the measured and modelled curves with the same vertical scale of electrons per Ry*. Although the match between experiment and theory is not exact, the overall agreement suggests that the donor-molecule model captures the correct physics. The dotted red curve addresses the discrepancy with regard to peak A, for which the predicted peak is significantly broader than the measurement; here we have reduced the broadening in the calculation by positioning the 2D layer 8 nm closer to the donor layer. In reality, reduced broadening may arise from increased screening in the donor layer.
Figure 3b shows schematically the full model we have developed. For simplicity, we assume a parallel-plate interaction between the tip and donor layer. As discussed in detail in Supplementary Information, Fig. S4, this simplification neglects a source of energy broadening that is significant, but less than other sources of broadening included explicitly in the model. The model considers a two-dimensional area of π(60 nm)2 with a fixed number of donors (labelled i in Fig. 3b) positioned randomly within the area. We then find the nearest neighbour for each donor to form a set of 2DMs (labelled k), each of which has assigned to it an addition spectrum ɛk1,2,3,4 based on the separation of the two atoms, as given by the configuration-interaction calculations (Fig. 3a). We account for the effects of non-nearest neighbours i by adding the Coulomb energy Uik due to all the other donors to every quantum level. The variation of this energy (each 2DM has a different configuration of neighbours) is the main source of broadening of the addition spectrum peaks. Last, the model includes the screening effect of the nearby 2D layer in our sample by using appropriately positioned image charges. The Methods section includes more details of the modelling procedure and a discussion of the contribution of 3DMs.
Figure 3c shows a representative measured capacitance curve and curves calculated with the full 2DM model. The measured curve was acquired at a single location; similarly, the modelled curves are two examples of individual random configurations of dopants within the simulated area. Figure 3d compares a measured curve which is the average of three locations with a model curve obtained by averaging over many random configurations of donors. Understanding the detailed shape of each broad peak that results from this calculation is subtle, stemming from the fact that the ionization of the system changes during the measurement; that is, the donors become neutralized as charge is added to the layer. The modelling shows that the resulting peak width is roughly 1 Ry*. To directly compare measurement with theory, the background step feature has been subtracted from the measured data, which are shown in units of effective Rydbergs and shifted horizontally so that peak C is at zero. This is consistent with peak C lying near zero effective Rydbergs, the energy above which the electrons are unbound. No other free parameters were used in the comparison.
We see that the charge magnitude and relative energy spacing of the model peaks are in good agreement with the measurements, with peaks A and B corresponding to the average addition energies of the first and second electron ɛ1,2 as indicated; the model predicts that the third and fourth electron peaks ɛ3,4 will be unresolved, consistent with peak C. Hence, we believe that the 2DM model captures the key physics of the system. However, the model does not account for all of the features in the data, in particular the smaller peaks indicated by the coloured arrows in Fig. 3d. With regard to the peak near −7 Ry*, indicated by the left-hand brown arrow, the energy and magnitude are approximately consistent with the expected first-electron peak for 3DMs; as discussed in the Methods section, a rough estimate gives −9 Ry* and 3.5 e/Ry* for the energy and magnitude of this peak. No other 3DM peaks are as prominent in the measured data, probably owing to the overlap between the 3DM and 2DM spectra. We believe that some of the measured structure between −6 Ry* and 0 Ry*, such as the small peak near −5 Ry* (right-hand brown arrow), arises from 3DMs. Small peaks also occur at positive energy, indicated by the purple arrows in Fig. 3d. In this case, we speculate that the peaks arise from the interplay between the tip potential and donor-molecule resonant states (that is, virtual bound states).
Figure 4 shows data acquired both in zero magnetic field and with 4.5 T applied in the z direction. We see that peaks A and B shift to lower energy by about −1 Ry*, whereas peak C shifts by only a small amount—but splits into two peaks. This same behaviour has been observed at three sample locations. To the best of our knowledge, no complete theory exists for the addition spectrum of hydrogenic molecules subject to a perpendicular magnetic field. However, we can compare our observation with the well-established behaviour of isolated atoms in magnetic field, such as isolated silicon donors in GaAs (refs 20, 21, 22). In the context of the donor-molecule interpretation of our data, the observed −1 Ry* shift of peaks A and B is consistent with the isolated Si studies in that it is similar to the behaviour of the one-electron (D0) and two-electron (D−) states. Physically, the dominant effect of the magnetic field is that it squeezes the ground state orbit closer to the positive ion(s), thus lowering the energy. This effect is expected to be reduced for less localized states such as peak C. Although there is no theory that predicts the details of the splitting of peak C, it is certainly consistent with our interpretation that it consists of two unresolved states at zero field.

The two curves were acquired at the same (single) location as the data shown in Fig. 3c, with the indicated magnetic field applied perpendicular to the donor plane. They are shown without subtracting the background capacitance; the grey lines serve as guides to the eye. The observed shift to lower energy of peaks A and B is consistent with the expected behaviour of strongly localized states, whereas peak C clearly splits into two peaks. This behaviour is generally consistent with the donor-molecule interpretation of the resonances. We have found that a small magnetic response of our piezoelectric scanning assembly shifts the position of the tip laterally on the scale of tens of nanometres. For this reason we are not probing exactly the same set of donors as the magnetic field increases. The curves were acquired with an excitation voltage amplitude of Vexc=15 mV r.m.s.
Methods
For the local probe measurements, we begin each data run by scanning the tip in both tunnelling and capacitance modes to check that the surface is sufficiently clean and free from major electronic defects13. To acquire the capacitance curves, we position the tip about 1 nm from the GaAs surface and hold it at a fixed location while sweeping the tip voltage. The peaks seen in the charging signal show little lagging-phase structure and can be considered as essentially a change in capacitance; in contrast, the background plateau structure shows a significant phase shift as large as π/8. These observations are detailed in Supplementary Information, Fig. S2, which also presents an equivalent circuit for the measurement. Our sensor circuit includes a bridge that enables us to subtract the background mutual capacitance of the tip and sample, ∼20 fF. Hence our plotted signal represents the change in capacitance as a function of voltage. All voltages are plotted with respect to the effective zero voltage. This is the voltage for which no electric field terminates on the top electrode (gate or tip); it is shifted from the applied voltage by an amount equal to the contact potential, Vcontact. For the PtIr tip used in the local probe measurements, Vcontact=0.60 V, as determined from in situ Kelvin probe measurements12. For the gated capacitance data, the observed shift in the curves implies Vcontact=0.12 V; this value agrees reasonably well with the reported work functions of Ti and Au, in comparison to Pt and Ir (ref. 23).
In general, single electrons can be resolved by capacitance techniques at helium temperatures if the energy spacing to add successive electrons is on the millivolt scale or greater. As described in detail in ref. 24, by measuring the capacitance C, we can detect charges entering the quantum system below the probe. We define the addition energy ɛn as the energy for which the nth electron enters the system. As Vtip increases from zero, the energy of an electron at the layer decreases as −αtipe Vtip, where αtip is the geometry-dependent proportionality constant, sometimes referred to as the voltage lever arm. In other words, electrons in the underlying 2D electrode are pulled toward the donor layer. The first electron will enter when the chemical potential is equal to the ground-state energy of the one-electron quantum state, ɛ1=E(1). As Vtip increases further, the second electron will be induced to enter when the chemical potential is equal to the energy difference between two-electron and one-electron ground states, ɛ2=E(2)−E(1). In general, ɛn=E(n)−E(n−1), where we define E(0)≡0. The capacitance C is given by C≡dq/dV ∝∂〈n〉/∂ μ, where dq corresponds to the tip charge, dV corresponds to the excitation voltage, μ is the chemical potential and 〈n〉 is the expectation value for the number of electrons in the system.
For our experiment, there are two lever-arm parameters. For the gated measurement shown in Fig. 2d, the parallel-plate geometry and sample growth parameters give a proportionality constant of 1/4.0 with respect to the donor layer. For the local probe measurements, the relative scale factor between the two voltage ranges used in Fig. 2d implies an effective lever arm of αtip=1/10.8. This is a reasonable value, consistent with the expected tip–sample mutual capacitance25,26,27.
With regard to our donor-molecule model, we consider a two-dimensional area with donors i positioned randomly. Each donor is paired with its nearest neighbour to form molecule k and an addition spectrum ɛk1,2,3,4 is assigned to it, based on the separation of the two atoms, as given by the configuration-interaction calculations (Fig. 3a). For a small fraction of donors, ambiguous cases can arise, such as equidistant nearest neighbours. In these cases the assignment of pairs is arbitrary. As discussed above, our model should apply to only about 38% of the donors, or roughly one-third. We account for this in a simple way: instead of considering 140 donors, the nominal number in the probed area of π(60 nm)2, the model considers only 140/3≈46. To accurately simulate the capacitance measurement, we must consider that the ionization of the system changes throughout the measurement, as shown schematically in Fig. 3b. For example, for the first electron to enter the area, we assume all the donors are ionized. Hence we calculate the Coulomb shifts for each pair k,  , using a charge of +e for all donors. In this case, the pair that has the lowest energy
, using a charge of +e for all donors. In this case, the pair that has the lowest energy  receives the electron, filling its first state and thus contributing to the capacitance at this energy. For all subsequent electron additions to other pairs, we must consider that this particular pair no longer has two fully ionized atoms. In other words, for the second electron, which would probably enter some other pair, the Coulomb shifts will be slightly reduced owing to the previous charge that has already entered the system and partially neutralized one pair of atoms. For simplicity, our modelling routine assumes perfect screening: every time an electron enters a 2DM, we add −e/2 to each atom of the pair. On average, the Coulomb shifts result in a broadening of the addition resonances ∼1 Ry*.
receives the electron, filling its first state and thus contributing to the capacitance at this energy. For all subsequent electron additions to other pairs, we must consider that this particular pair no longer has two fully ionized atoms. In other words, for the second electron, which would probably enter some other pair, the Coulomb shifts will be slightly reduced owing to the previous charge that has already entered the system and partially neutralized one pair of atoms. For simplicity, our modelling routine assumes perfect screening: every time an electron enters a 2DM, we add −e/2 to each atom of the pair. On average, the Coulomb shifts result in a broadening of the addition resonances ∼1 Ry*.
The modelling method accounts for isolated donors in an approximate way. For example, suppose the nearest neighbour of a particular donor is 10a0*; the routine will still pair the donors and assign an addition spectrum. However, the addition spectrum of two donors with such a large separation is approximately equal to the addition spectrum of isolated donors. This can be seen in Supplementary Information, Fig. S3c (which neglects the tip potential): at large separation E(1) tends to −1.0 Ry, whereas E(2) tends to −2.0 Ry. Therefore both ɛ1=E(1) and ɛ2=E(2)−E(1) are approximately equal to −1.0 Ry for very large separations. This corresponds to two electrons entering the ‘molecule’ at the same energy, equal to that of the isolated ion.
The model assumes that the dopants are perfectly confined to a plane, thus neglecting the effects of Si migration during molecular-beam epitaxial growth28. For our sample, the z distribution of the Si is expected to have a half-width at half-maximum of 6 nm, comparable to the effective Bohr radius29. The most significant consequence of neglecting this effect is that the model underestimates the nearest-neighbour distances d by roughly a0*. However, as can be seen in Fig. 3a, a change in d of a0* results in a variation of addition energies of less than 1 Ry*. Hence this contribution to the energy broadening is less than the Coulomb-shift broadening effect of the non-nearest neighbours, which is included in the model.
We have used our donor molecule model to generate examples of capacitance curves corresponding to single locations, as shown in Fig. 3c, and to generate the average characteristic structure by repeating the procedure for hundreds of random ensembles and averaging the results, as shown in Fig. 3d. We see that, on average, the model predicts three peaks, which correspond reasonably well to peaks A, B and C. The reason the model gives only three peaks despite the fact that there are four electrons per molecule is simple: ɛ3 and ɛ4 are separated by roughly 0.5 Ry; hence, they are not resolvable as individual peaks. The reason the peaks have distinct shapes is more subtle, arising from the ionization effects described above. For example, for the second electron additions ɛ2, on average there are fewer ionized charges in the donor layer than for the first electron additions ɛ1. Therefore the overall Coulomb shift is reduced for the second electrons, which form peak B, as well as the broadening effect due to the randomness in the donor positions. For this reason the model predicts that, on average, peak B will be sharper than peak A. The shape of peak C is also broadened by the proximity of ɛ3 and ɛ4.
The model does not account for clusters of three or more donors. Among these larger clusters, the contribution of three-donor molecules should be most prominent; as discussed above, on average 12 3DM should be present in the probed area, compared to 26 2DMs (and only six four-donor molecules). Although we have not developed a simulation to account for 3DMs, we can use the results of the 2DM model as a rough guide for the behaviour of the 3DM first electron additions. For both 2DMs and 3DMs, the first electron addition ɛ1 can be estimated using the expression ɛ1=−Z2, where Z is the nuclear charge of the ions and the units of energy are Ry*; this is exact in the limit of zero separation of the ions within the molecule, in which case we have a hydrogenic potential (and no electron–electron interactions to consider as this is the first electron). The expression gives ɛ12DM=−22=−4 Ry*, in surprisingly good agreement with the average first-electron peak calculated by the full 2DM model, as shown in Fig. 3d. The agreement results from an approximate cancellation of three effects: on average the two molecules are separated by 1.2a0*, which tends to increase ɛ1; however, the Coulomb shift from non-nearest neighbours and the tip potential tend to decrease ɛ1. These results imply that we should expect, on average, ɛ13DM≈−32=−9 Ry*. With regard to the magnitude of the 3DM first-electron peak, we expect it to scale approximately with the number of molecules. Figure 3d shows the calculated magnitude of the 2DM peak to be 7.5 e/Ry*; hence, we expect the first 3DM peak to be ∼(12/26)7.5 e/Ry*=3.5 e/Ry*.
References
Kane, B. E. A silicon-based nuclear spin quantum computer. Nature 393, 133–137 (1998).
Vrijen, R. et al. Electron-spin-resonance transistors for quantum computing in silicon–germanium heterostructures. Phys. Rev. A 62, 012306 (2000).
Hollenberg, L. C. L. et al. Charge-based quantum computing using single donors in semiconductors. Phys. Rev. B 69, 113301 (2004).
Kohn, W. & Luttinger, J. M. Theory of donor states in silicon. Phys. Rev. 98, 915–922 (1955).
Lieb, E. H. Bound on the maximum negative ionization of atoms and molecules. Phys. Rev. A 29, 3018–3028 (1984).
Ruess, F. J. et al. Realization of atomically controlled dopant devices in silicon. Small 3, 563–567 (2007).
Andresen, S. E. S. et al. Charge state control and relaxation in an atomically doped silicon device. Nano Lett. 7, 2000–2003 (2007).
Ashoori, R. C. Electrons in artificial atoms. Nature 379, 413–419 (1996).
Deshpande, M. R., Sleight, J. W., Reed, M. A., Wheeler, R. G. & Matyi, R. J. Spin splitting of single 0D impurity states in semiconductor heterostructure quantum wells. Phys. Rev. Lett. 76, 1328–1331 (1996).
Sellier, H. et al. Transport spectroscopy of a single dopant in a gated silicon nanowire. Phys. Rev. Lett. 97, 206805 (2006).
Geim, A. K. et al. Resonant tunneling through donor molecules. Phys. Rev. B 50, 8074–8077 (1994).
Tessmer, S. H., Glicofridis, P. I., Ashoori, R. C., Levitov, L. S. & Melloch, M. R. Subsurface charge accumulation imaging of a quantum Hall liquid. Nature 392, 51–54 (1998).
Urazhdin, S., Maasilta, I. J., Chakraborty, S., Moraru, I. & Tessmer, S. H. High-scan-range cryogenic scanning probe microscope. Rev. Sci. Instrum. 71, 4170–4173 (2000).
Ashoori, R. C. et al. Single-electron capacitance spectroscopy of discrete quantum levels. Phys. Rev. Lett. 68, 3088–3091 (1992).
Schmidt, T., Muller, St. G., Gulden, K. H., Metzner, C. & Dohler, G. H. In-plane transport properties of heavily d-doped GaAs n–i–p–i superlattices: Metal–insulator transition, weak and strong localization. Phys. Rev. B 54, 13980–13995 (1996).
Slater, J. C. Quantum Theory of Molecules and Solids Vol. 1 Electronic Structure of Molecules (McGraw-Hill, New York, 1963).
Dunning, T. H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 90, 1007–1023 (1989).
Davies, J. H. The Physics of Low-dimensional Semiconductors: An Introduction (Cambridge Univ. Press, Cambridge, 1998).
Levinstein, M., Rumyantsev, S. & Shur, M. Handbook Series on Semiconductor Parameters Vol. 2 (World Scientific, London, 1999).
Lok, J. G. S. et al. D− centers probed by resonant tunneling spectroscopy. Phys. Rev. B 53, 9554–9556 (1996).
Shi, J. M., Peeters, F. M. & Devreese, J. T. D− states in GaAs/AlxGa1−xAs superlattices in a magnetic field. Phys. Rev. B 51, 7714–7724 (1995).
Shi, J. M., Peeters, F. M., Hai, G. Q. & Devreese, J. T. Donor transition energy in GaAs superlattices in a magnetic field along the growth axis. Phys. Rev. B 44, 5692–5702 (1991).
Lide, D. R. CRC Handbook of Chemistry and Physics 86th edn (Taylor & Francis, Boca Raton, 2005–2006).
Kaplan, T. A. The chemical potential. J. Stat. Phys. 122, 1237–1260 (2006).
Eriksson, M. A. et al. Cryogenic scanning probe characterization of semiconductor nanostructures. Appl. Phys. Lett. 69, 671–673 (1996).
Kuljanishvili, I., Chakraborty, S., Maasilta, I. J. & Tessmer, S. H. Modeling electric field sensitive scanning probe measurements for a tip of arbitrary shape. Ultramicroscopy 102, 7–12 (2004).
Tessmer, S. H., Finkelstein, G., Glicofridis, P. I. & Ashoori, R. C. Modeling subsurface charge accumulation images of a quantum Hall liquid. Phys. Rev. B 66, 125308 (2002).
Mills, A. P. Jr, Pfeiffer, L. N., West, K. W. & Magee, C. W. Mechanisms for Si dopant migration in molecular beam epitaxy AlxGa1−xAs. J. Appl. Phys. 88, 4056–4060 (2000).
Pfeiffer, L. N., Schubert, E. F., West, K. W. & Magee, C. W. Si dopant migration and the AlGaAs/GaAs inverted interface. Appl. Phys. Lett. 58, 2258–2260 (1991).
Acknowledgements
We gratefully acknowledge helpful comments and advice from R. C. Ashoori, N. Birge, M. I. Dykman, B. Golding, S. D. Mahanti, I. J. Maasilta, S. Rogge and G. A. Steele. This work was supported by the Michigan State Institute for Quantum Sciences and the National Science Foundation, grant Nos DMR-0305461 and DMR-0605801.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Information
Supplementary Figure 1–4 (PDF 252 kb)
Rights and permissions
About this article
Cite this article
Kuljanishvili, I., Kayis, C., Harrison, J. et al. Scanning-probe spectroscopy of semiconductor donor molecules. Nature Phys 4, 227–233 (2008). https://doi.org/10.1038/nphys855
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nphys855
This article is cited by
-
Atom devices based on single dopants in silicon nanostructures
Nanoscale Research Letters (2011)
-
Single dopants in semiconductors
Nature Materials (2011)
-
Trionic optical potential for electrons in semiconductors
Nature Physics (2010)
-
Probing dopants at the atomic level
Nature Physics (2008)
-
A close look at doping
Nature Nanotechnology (2008)
