
Abstract
The application of optical forces has allowed unprecedented control over the motion of atoms leading to laser cooling and trapping1 and Bose–Einstein condensation2. More recently, the manipulation of cold neutral polar molecules using electrostatic fields has been used to create slow cold molecules in the laboratory frame3. Here, we report on the controlled manipulation of molecules using deep periodic optical lattice potentials (22 K) created by intense optical fields (1011 W cm−2). By using the oscillatory motion of nitric oxide molecules in these potentials, we have demonstrated rapid deceleration or acceleration in a molecular beam to well-defined velocities on a length scale of less than 535 nm. We reduce the kinetic energy of the molecules in a cold molecular beam by up to 50% in a single laser pulse (5.8 ns) demonstrating precise control of molecular-beam velocity and a new method for creating slow cold molecules using optical fields.
Similar content being viewed by others
Main
New techniques for creating cold stationary molecules are actively being pursued because, in general, molecules cannot be laser cooled4,5,6,7,8. An important subset of these techniques uses electrostatic potentials formed by the interaction between an applied electric field and the dipole moment of a molecule. These potentials are used to trap and slow a group of cold molecules in a molecular beam bringing them to rest3,9. An optical Stark potential, which relies on the interaction between the field and an induced dipole moment, can also be used to manipulate and slow molecules10,11,12,13,14,15,16. In this letter, we describe the measurement of rapid deceleration or acceleration of nitric oxide (NO) molecules that are trapped within deep periodic optical lattice potentials moving at constant velocity. To the best of our knowledge this is the first report of the observation of the rapid deceleration and acceleration of molecules in an optical lattice that is based on previous theoretical studies17. We show that for a fixed-duration optical pulse, the velocity of the trapped molecules can be determined by the difference between the molecular-beam and lattice velocity over a wide range (∼250 m s−1). Importantly, the velocity spread of the trapped molecules transported by the lattice is approximately the same as the unperturbed distribution function.
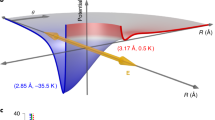
A one-dimensional optical lattice is a periodic array of potential wells created by the interaction between a polarizable particle and an interference pattern created by two near-counterpropagating laser beams as shown in Fig. 1. The slowly varying lattice potential is given by  , where ɛ0 is the permittivity of free space, c is the speed of light, I1(t) and I2(t) are the intensities of each optical field, α is the effective polarizablility, Δω is the frequency difference between the two lattice beams, and k is the lattice wavenumber given by k=(4πsinφ)/λ where λ is the mean wavelength of the lasers and φ is the half angle between the two fields17,18,19. A fixed frequency difference between the two pump fields produces a lattice moving with velocity vL=Δω/k in the laboratory frame. The oscillatory motion that occurs along the axis of the constant velocity lattice can be described by a phase-space plot in a reference frame (yr,vr) that moves with the lattice velocity (ref. 17), shown in Fig. 2, where vr=v−vL is the molecule's velocity with respect to the lattice velocity and yr=y−vLt. The lines with arrows in this figure represent the trajectories of NO molecules in the (yr,vr) plane with an initial velocity of 400 m s−1 and a lattice velocity of 321 m s−1. This figure is produced assuming a constant well depth of U/kB=22 K, where kB is Boltzmann's constant, and I1=I2=2.2×1011 W cm−2, α=1.9×10−40 C m2 V−1, k=1.15×107 m−1. The arrows in Fig. 2 indicate the direction that molecules move, following lines of equal energy, and the thick solid lines represent the separatrix that divide the phase space into two regions. Inside, the molecules are trapped within the lattice potential, whereas outside they are untrapped but can still be significantly perturbed. As the potential is periodic, the dynamics in any lattice site are repeated every lattice period along the y axis. The horizontal blue line represents the initial velocities and positions of molecules that are in a cold molecular beam (1.8 K) before the lattice is turned on. These molecules have an initial velocity that is 79 m s−1 greater than the lattice velocity. The trajectories of trapped molecules at two different locations along the y axis are shown by A→A′ and B→B′, which represent the motion of two trapped molecules in a molecular beam that undergo a half oscillation within the potential well when it is turned on for a duration of 6 ns. This is referred to as a half rotation in the phase space. Theoretical studies have shown that a half rotation in the optical lattice is optimal for creating a bunch of decelerated or accelerated molecules with approximately the same velocity spread as the original beam17. A half rotation in the trapped region of Fig. 2 results in molecules decelerated to a velocity that is 79 m s−1 less than the lattice velocity when the field is turned on for the time required for the half rotation (6.0 ns). In the laboratory frame this corresponds to a deceleration from 400 m s−1 to a final velocity of 242 m s−1. If the lattice velocity is greater than the molecular-beam velocity, a half rotation results in the acceleration of molecules to a velocity that is greater than the lattice. Thus, using a half rotation, the relative velocity of decelerated or accelerated molecules is approximately twice the difference between the lattice and molecular-beam velocity. However, owing to the anharmonicity of the potential, the time for a half phase-space rotation increases with the magnitude of the velocity difference between the lattice and the molecular beam.
, where ɛ0 is the permittivity of free space, c is the speed of light, I1(t) and I2(t) are the intensities of each optical field, α is the effective polarizablility, Δω is the frequency difference between the two lattice beams, and k is the lattice wavenumber given by k=(4πsinφ)/λ where λ is the mean wavelength of the lasers and φ is the half angle between the two fields17,18,19. A fixed frequency difference between the two pump fields produces a lattice moving with velocity vL=Δω/k in the laboratory frame. The oscillatory motion that occurs along the axis of the constant velocity lattice can be described by a phase-space plot in a reference frame (yr,vr) that moves with the lattice velocity (ref. 17), shown in Fig. 2, where vr=v−vL is the molecule's velocity with respect to the lattice velocity and yr=y−vLt. The lines with arrows in this figure represent the trajectories of NO molecules in the (yr,vr) plane with an initial velocity of 400 m s−1 and a lattice velocity of 321 m s−1. This figure is produced assuming a constant well depth of U/kB=22 K, where kB is Boltzmann's constant, and I1=I2=2.2×1011 W cm−2, α=1.9×10−40 C m2 V−1, k=1.15×107 m−1. The arrows in Fig. 2 indicate the direction that molecules move, following lines of equal energy, and the thick solid lines represent the separatrix that divide the phase space into two regions. Inside, the molecules are trapped within the lattice potential, whereas outside they are untrapped but can still be significantly perturbed. As the potential is periodic, the dynamics in any lattice site are repeated every lattice period along the y axis. The horizontal blue line represents the initial velocities and positions of molecules that are in a cold molecular beam (1.8 K) before the lattice is turned on. These molecules have an initial velocity that is 79 m s−1 greater than the lattice velocity. The trajectories of trapped molecules at two different locations along the y axis are shown by A→A′ and B→B′, which represent the motion of two trapped molecules in a molecular beam that undergo a half oscillation within the potential well when it is turned on for a duration of 6 ns. This is referred to as a half rotation in the phase space. Theoretical studies have shown that a half rotation in the optical lattice is optimal for creating a bunch of decelerated or accelerated molecules with approximately the same velocity spread as the original beam17. A half rotation in the trapped region of Fig. 2 results in molecules decelerated to a velocity that is 79 m s−1 less than the lattice velocity when the field is turned on for the time required for the half rotation (6.0 ns). In the laboratory frame this corresponds to a deceleration from 400 m s−1 to a final velocity of 242 m s−1. If the lattice velocity is greater than the molecular-beam velocity, a half rotation results in the acceleration of molecules to a velocity that is greater than the lattice. Thus, using a half rotation, the relative velocity of decelerated or accelerated molecules is approximately twice the difference between the lattice and molecular-beam velocity. However, owing to the anharmonicity of the potential, the time for a half phase-space rotation increases with the magnitude of the velocity difference between the lattice and the molecular beam.

The ultraviolet probe beam that was used to ionize and detect the NO molecules perturbed by the lattice is also shown. The velocity of the ions, and thus that of the neutral molecules, was determined from their time-of-flight in a mass spectrometer.

The lines with arrows represent the trajectory of molecules of equal energy, and the arrows indicate the direction of this motion. A molecular beam travelling at the velocity of the unperturbed jet is represented by the horizontal blue line. The trajectories A→A′ and B→B′ indicate the initial and final location and velocity of two molecules that undergo a half phase-space rotation, within the trapped region when the 22-K deep optical lattice potential is turned on for 6.0 ns.
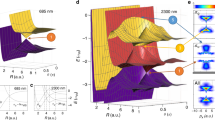
To verify that essentially the same dynamics occur when using a realistic pulsed lattice with nanosecond turn-off and turn-on times, we have simulated the motion of an ensemble of NO molecules at 1.8 K initially travelling at an average velocity of 400 m s−1. The velocity distribution function of the ensemble was calculated as a function of time by numerical integration of the one-dimensional Boltzmann equation19, for an average well depth of 22 K, using experimentally measured intensity profiles produced by the two Q-switched Nd:YAG lasers. The temporal lattice intensity profile produced by separate measurements of the two Q-switched Nd:YAG laser profile used to create the optical lattice is shown in Fig. 3a. Figure 3b is a simulation of the evolution of the velocity distribution function for the duration of the pulsed lattice. For this pulse duration the molecules undergo an oscillation that is slightly greater than a half rotation because of the finite turn-off time of the potential. They are decelerated by 130 m s−1, whereas a half rotation achieved with a potential that is rapidly switched on a faster timescale than the dynamics would result in a velocity reduction of 158 m s−1.

a, The temporal lattice intensity profile produced by separate measurements of the two Q-switched Nd:YAG laser profiles used to create the optical lattice. b, The simulation of the dynamics of molecules within a constant-velocity optical lattice using the lattice profile of a. A lattice moving at 321 m s−1 (solid white line) with an average well depth of 22 K was used to decelerate a portion of NO molecules from 400 to 270 m s−1 representing a near-half rotation in phase space. The white dotted lines represent the extent of the time-dependent trapped region within the lattice potential; the colours represent the relative molecular density.
We measure the dynamics of cold (1.8 K) NO molecules initially travelling at 400 m s−1 within a molecular beam, perturbed by an optical lattice with an average well depth of 22 K and 5.8-ns duration, as shown in Fig. 1. To probe the motion of the molecules after the lattice was turned off, they were ionized by a resonantly enhanced multiphoton ionization (REMPI) scheme. The velocity of the ions, and thus the neutrals along the y axis, was determined by their time-of-flight in a mass spectrometer20. Figure 4 shows plots of the measured velocity distribution of molecules in the lattice for a range of lattice velocities. Each plot is labelled with the lattice velocity relative to the molecular-beam velocity. Adjacent to the experimental plots are simulations of the velocity distribution for each lattice velocity. For comparison, the unperturbed velocity distribution functions are also shown. For lattice velocities less than the molecular-beam velocity (−79,−52,−26 m s−1), a significant fraction of the probed molecules are decelerated, and conversely, for lattice velocities greater than the molecular-beam velocity (52, 26 m s−1), molecules are accelerated, with approximately the same velocity spread as the initial unperturbed molecular beam. The large fraction of molecules that remain near the initial molecular-beam velocity are mostly due to the detection of unperturbed molecules outside the lattice. As the magnitude of the relative velocity increases, a smaller fraction of molecules are accelerated or decelerated because less are confined within the separatrix or trapped region shown in Fig. 2. Our simulations show all of the major features measured in the experiment, indicating that most trapped molecules undergo approximately a half oscillation (half rotation in phase space) for all the relative velocities studied. We believe the differences between the exact peak separations and location in the experiment and the simulations in Fig. 4 can be attributed to the uncertainty in determining the intensity (20%) of each of the lattice beams and the velocity of the molecular beam (3%). The uncertainty in intensity results primarily from a 10% uncertainty in the energy and a 5% uncertainty in our measurements of laser beam waist. There is also a 10% uncertainty in the lattice-beam spatial overlap. The difference between our results and the idealized half rotation represented in Fig. 2 was attributed to the detection of molecules in the x–z plane of Fig. 1 which have been perturbed by the radially varying lattice potential, which is not as deep as the on-axis potential, and thus the molecules do not undergo a half space rotation. In addition, for the different lattice velocities that we used, there is approximately a 10% variation in the time required for a half phase-space rotation for molecules that are on-axis due to the anharmonicity of the lattice potential. Figure 4 shows that approximately 7% of the NO molecules in the detection volume are decelerated to 270 m s−1 with a lattice velocity of 321 m s−1. However, our simulations indicate that the majority of molecules (75%) in the central on-axis region (20 μm) of the lattice undergo a half rotation in phase space and are accelerated/decelerated to the desired velocity. Our REMPI measurements indicate a lower bound to the density of 5×1010 cm−3, which corresponds to a slowed number of 2×105 molecules per pulse. This number can be increased by at least an order of magnitude by carrying out experiments closer to the nozzle from which the molecular beam originates. The lowest and highest velocities that can be achieved by the half rotation are determined by the lattice intensity and pulse duration at which significant ionization or dissociation occur.

Lattice velocities with respect to the molecular-beam velocity of −79,−52,−26, 0, 26 and 52 m s−1 are shown. For comparison, simulations of the perturbed distribution function created by the different velocity lattices is shown alongside the experimental data.
Our measurements have demonstrated controlled acceleration and deceleration of NO molecules using a half oscillation within a deep optical lattice moving at constant velocity. The NO molecules experience an average deceleration/acceleration of over 109 g over the 6-ns period of this motion and a maximum kinetic energy change of up to 50% for an average well depth of 22 K. Although we have demonstrated this process with NO molecules, this is a general method, which can be applied to essentially any species in a cold molecular beam. By controlling lattice velocity, well depth and duration, any molecular velocity over a wide range can be produced and could be used to create slow cold molecules in the laboratory frame.
Methods
Simulations of molecular dynamics in the optical lattice
To simulate the dynamics of the molecules in the time-dependent lattice potential we solved the one-dimensional, non-stationary, collisionless Boltzmann equation with periodic boundary conditions using the second-order explicit MacCormack method with flux-corrected transport18,19,21,22.
Molecular-beam and lattice creation
A cold molecular beam of NO was created at a temperature of approximately 1.8 K by expanding xenon containing 5% by volume of NO at a total pressure of 1.5 atmospheres through a pulsed valve into a vacuum chamber. This beam was collimated by a skimmer and passed into a second differentially pumped chamber with an average pressure of 1×10−7 torr. The molecular beam intersected the pulsed lattice at a distance of approximately 300 mm from the orifice of the pulsed valve. The lattice was produced by two, injection seeded, Q-switched, Nd:YAG lasers operating in the infrared at a wavelength of 1,064 nm, producing an average laser intensity of 2.2×1011 W cm−2. The effective polarizability for a molecule is given by  , where α|| and
, where α|| and  are the polarizability components parallel and perpendicular to the molecular axis, and θ is the angle between the molecular axis and the laser polarization. The effective polarizability of NO is α=1.9×10−40 C m2 V−1, which at this intensity leads to an average lattice well depth of 22 K.
are the polarizability components parallel and perpendicular to the molecular axis, and θ is the angle between the molecular axis and the laser polarization. The effective polarizability of NO is α=1.9×10−40 C m2 V−1, which at this intensity leads to an average lattice well depth of 22 K.
The frequencies of the two infrared lasers were adjusted to produce an optical lattice of constant velocity. The frequency difference between the two infrared lasers was determined by heterodyning each pulsed laser beam with a stable continuous-wave reference laser with a linewidth of 10 kHz. We vary the frequency of one laser while the other is fixed. This frequency is controlled by a variation in temperature of the seeder laser. A decrease in the temperature results in an increase in frequency (1.4 GHz/degree). If we increase the frequency of the laser, and the beat frequency increases, then we know that the laser frequency is above that of the fixed frequency laser. Similarly, if we increase the frequency and beat frequency decreases, we know that we are below the frequency of the fixed frequency laser. In this way we can determine the sign of the frequency difference. This frequency difference was measured as a function of time during the pulse, and was used to determine the instantaneous lattice velocity. Each lattice beam was focused to a spot size with a full-width at half-maximum of 60 μm and crossed at an angle of 167.5∘, creating a lattice with a period of approximately 535 nm.
Measurement of molecular velocities
After the molecules were accelerated or decelerated by the optical lattice, they were ionized by a single-colour (1+1) REMPI process from the lowest vibrational (v′′=0) and rotational (j′′=1/2) state of the electronic ground state X2Π1/2 through the A2Σ+ state using 226-nm light from the frequency doubled output from a dye laser operating on Coumarin 450 laser dye23. The probe beam was focused to an estimated beam waist of approximately 2 μm in the x–y plane of Fig. 1. The lattice and probe beams were focused inside a Wiley–McLaren type mass spectrometer (not shown in the figure), and their velocity was determined by their time-of-flight within the spectrometer20. The perturbed molecules were ionized 60 ns after the peak lattice intensity. This delay between the lattice beams and the probe was required because the long tail of the infrared pulses produces a field that can distort the REMPI process due to the a.c. Stark shift of the intermediate excited state. We estimate that ion recoil due to the ionization process introduces less than 2% uncertainty in the measurement of the velocity of the neutral NO molecules. The Rayleigh range of the focused ultraviolet probe beam is larger than the diameter of the focused beams that form the lattice, and therefore molecules that are outside this region are ionized by the probe and are also detected. To reduce detection of these unperturbed molecules, a slit of width 150 μm was placed over the entrance of the drift tube of the spectrometer. All measurements of the velocity distribution function were an average of 1,000 lattice pulses.
References
Kasevich, M. & Chu, S. Laser cooling below a photon recoil with three-level atoms. Phys. Rev. Lett. 69, 1741–1745 (1992).
Anderson, M. H., Ensher, J. R., Matthews, M. R., Wieman, C. E. & Cornell, E. A. Observation of Bose-Einstein condensation in a dilute atomic vapor. Science 269, 198–201 (1995).
Bethlem, H. L. et al. Electrostatic trapping of ammonia molecules. Nature 406, 491–494 (2000).
Schlöter, U., Silber, C. & Zimmermann, C. Photoassociation of heteronuclear lithium. Appl. Phys. B 73, 801–805 (2001).
Regal, C. A., Ticknor, C., Bohn, J. L. & Jin, D. S. Creation of ultracold molecules from a Fermi gas of atoms. Nature 424, 47–50 (2003).
Weinstein, J. D., deCarvalho, R., Guillet, T., Friedrich, B. & Doyle, J. M. Magnetic trapping of calcium monohydride molecules at millikelvin temperatures. Nature 395, 148–150 (1998).
Gupta, M. & Herschbach, D. Slowing and speeding molecular beams by means of a rapidly rotating source. J. Phys. Chem. A 105, 1626–1637 (2001).
Elioff, M. S., Valentini, J. J. & Chandler, D. W. Subkelvin cooling NO molecules via `billiard-like' collisions with argon. Science 302, 1940–1943 (2003).
Tarbutt, M. R. et al. Slowing heavy, ground-state molecules using an alternating gradient decelerator. Phys. Rev. Lett. 92, 173002 (2004).
Stapelfeldt, H., Sakai, H., Constant, E. & Corkum, P. B. Deflection of neutral molecules using the nonresonant dipole force. Phys. Rev. Lett. 79, 2787–2790 (1997).
Zhao, B. S. et al. Molecular lens of the nonresonant dipole force. Phys. Rev. Lett. 85, 2705–2708 (2000).
Zhao, B. S. et al. Separation of a benzene and nitric oxide mixture by a molecule prism. J. Chem. Phys. 119, 8905–8909 (2003).
Villeneuve, D. M. et al. Forced molecular rotation in an optical centrifuge. Phys. Rev. Lett. 85, 542–545 (2000).
Sakai, H. et al. Controlling the alignment of neutral molecules by a strong laser field. J. Chem. Phys. 110, 10235–10238 (1999).
Fulton, R., Bishop, A. I. & Barker, P. F. Focusing ground-state xenon in a pulsed optical field. Phys. Rev. A 71, 043404 (2005).
Fulton, R., Bishop, A. I. & Barker, P. F. Optical Stark decelerator for molecules. Phys. Rev. Lett. 93, 243004 (2004).
Dong, G., Lu, W. & Barker, P. F. Decelerating and bunching molecules with pulsed traveling optical lattices. Phys. Rev. A 69, 013409 (2004).
Barker, P. F. & Shneider, M. N. Slowing molecules by optical microlinear deceleration. Phys. Rev. A 66, 065402 (2002).
Barker, P. F. & Shneider, M. N. Optical microlinear accelerator for molecules and atoms. Phys. Rev. A 64, 033408 (2001).
Wiley, W. C. & McLaren, I. H. Time-of-flight mass spectrometer with improved resolution. Rev. Sci. Instrum. 26, 1150–1157 (1955).
Anderson, J. D. Jr Computational Fluid Mechanics (McGraw-Hill, New York, 1995).
Boris, J. P. & Book, D. L. Flux-corrected transport. 3. Minimal-error FCT algorithms. J. Comput. Phys. 20, 397–431 (1976).
Paul, P. H. Calculation of transition frequencies and rotational line strengths in the gamma-bands of nitric oxide. J. Quantum Spectrosc. Radiat. 57, 581–589 (1997).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Fulton, R., Bishop, A., Shneider, M. et al. Controlling the motion of cold molecules with deep periodic optical potentials. Nature Phys 2, 465–468 (2006). https://doi.org/10.1038/nphys339
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nphys339
This article is cited by
-
Raman gas self-organizing into deep nano-trap lattice
Nature Communications (2016)
-
Pushing atoms with light
Nature Photonics (2012)
-
Laser-driven acceleration of neutral particles
Nature Photonics (2012)
-
Superconducting microwave cavity towards controlling the motion of polar molecules
Applied Physics B (2012)
-
A high-energy, chirped laser system for optical Stark deceleration
Applied Physics B (2011)
