
Volume 8 Issue 11, November 2011
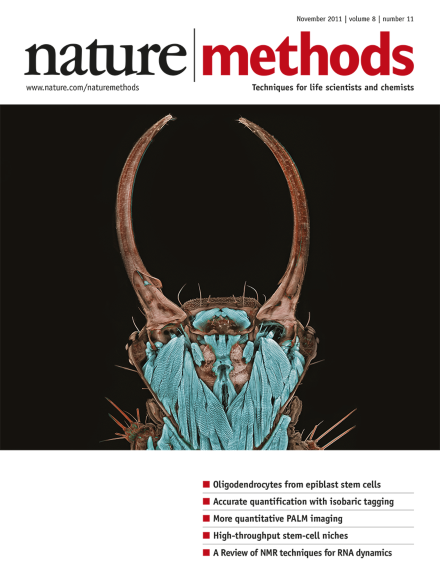
This photograph of a green lacewing larva (Chrysopa sp.) took first place in the 2011 Nikon Small World Photomicrography Competition. The image was taken by Igor Siwanowicz of the Max Planck Institute of Neurobiology, using confocal microscopy at 20× magnification. The full gallery of winning images can be viewed at http://www.nikonsmallworld.com/.
Editorial
-
Advertisement
