
Abstract
Sample heterogeneity often masks DNA methylation signatures in subpopulations of cells. Here, we present a method to genotype single cells while simultaneously interrogating gene expression and DNA methylation at multiple loci. We used this targeted multimodal approach, implemented on an automated, high-throughput microfluidic platform, to assess primary lung adenocarcinomas and human fibroblasts undergoing reprogramming by profiling epigenetic variation among cell types identified through genotyping and transcriptional analysis.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


References
Navin, N. et al. Nature 472, 90–94 (2011).
Zong, C., Lu, S., Chapman, A.R. & Xie, X.S. Science 338, 1622–1626 (2012).
Tang, F. et al. Cell Stem Cell 6, 468–478 (2010).
Patel, A.P. et al. Science 344, 1396–1401 (2014).
Guo, H. et al. Genome Res. 23, 2126–2135 (2013).
Smallwood, S.A. et al. Nat. Methods 11, 817–820 (2014).
Angermueller, C. et al. Nat. Methods 13, 229–232 (2016).
Hou, Y. et al. Cell Res. 26, 304–319 (2016).
Hu, Y. et al. Genome Biol. 17, 88 (2016).
Macaulay, I.C. et al. Nat. Methods 12, 519–522 (2015).
Bock, C., Farlik, M. & Sheffield, N.C. Trends Biotechnol. 34, 605–608 (2016).
Lorthongpanich, C. et al. Science 341, 1110–1112 (2013).
Cheow, L.F., Quake, S.R., Burkholder, W.F. & Messerschmidt, D.M. Nat. Protoc. 10, 619–631 (2015).
Kim, K. et al. Nature 467, 285–290 (2010).
Nishino, K. et al. PLoS Genet. 7, e1002085 (2011).
Selamat, S.A. et al. Genome Res. 22, 1197–1211 (2012).
Heller, G. et al. Carcinogenesis 34, 513–521 (2013).
Kerr, K.M., Galler, J.S., Hagen, J.A., Laird, P.W. & Laird-Offringa, I.A. Dis. Markers 23, 5–30 (2007).
Masters, G.A. et al. J. Clin. Oncol. 33, 3488–3515 (2015).
Rauch, T. et al. Proc. Natl. Acad. Sci. USA 104, 5527–5532 (2007).
Onder, T.T. et al. Nature 483, 598–602 (2012).
Sanchez-Freire, V., Ebert, A.D., Kalisky, T., Quake, S.R. & Wu, J.C. Nat. Protoc. 7, 829–838 (2012).
Acknowledgements
We thank T.H. Charn and S. Samydurai for technical help with developing Fluidigm C1 Scripts, C. El Farran for bioinformatics help, H.F. Wang for help with cell culture, and the GIS-Fluidigm Single-Cell Omics Center and the IMCB DNA Sequencing Facility for core facilities support. This work was supported by Career Development grant 14302FG095 (L.F.C. and E.T.C.) and an A*STAR Investigatorship grant (Y.-H.L.) from the Joint Council Office of the Agency of Science, Technology and Research. S.R.Q. is an investigator of the Howard Hughes Medical Institute.
Author information
Authors and Affiliations
Contributions
L.F.C. conceived the study. E.T.C., Y.-H.L., S.R.Q. and W.F.B. assisted in experimental design. L.F.C., E.T.C., Y.T., R.V., Q.X. and R.Z.T. performed the experiments and analyzed the data. D.S.W.T., P.R., Y.-H.L., S.R.Q. and W.F.B. supervised the study. L.F.C. wrote the manuscript. E.T.C., Y.T., R.V. and Q.X. contributed equally to the study. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
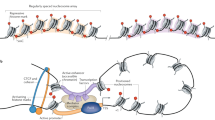
Supplementary Figure 1 Workflow for multimodal analysis using sc-GEM on a programmable microfluidic device (Fluidigm).
1) Cells are captured and lysed, 2) RNA from lysed single cell is reverse-transcribed to obtain complementary DNA (cDNA), 3) genomic DNA (gDNA) is digested with a methylation-sensitive restriction enzyme, 4) multiplexed PCR using primers specific for target cDNA (for gene expression assays) and target gDNA (for DNA methylation and genotype assays) is performed on-chip. The products from the chip are then collected, and a portion of each sample is reamplified with genotyping primers and tagged with sample-specific barcoded sequencing adapters. Multiple amplicons are pooled together and sequenced on a next generation sequencing platform to obtain the targeted genotype of the corresponding single cells. Another portion of each product collected from the chip is amplified with nested primers in a microfluidics qPCR chip (the Fluidigm Dynamic Array IFC) to obtain gene expression data and the DNA methylation status of the target loci. 5mC: Methylated CpG sites, blue strands: cellular RNA, red strands: cellular DNA, green strands: complementary DNA, orange strands: primers.
Supplementary Figure 2 Quality metrics of sc-GEM data obtained from unreprogrammed and reprogrammed human fibroblast.
(a-f) Sensitivity of sc-GEM gene expression detection was measured on bulk RNA samples, using a 1:1 mixture of RNA from fibroblast (BJ) and human ES cells. Titration curves on 10-fold dilution of bulk RNA showed that sc-GEM is able to amplify multiple target mRNA over a wide dynamic range. A parallel analysis on single cells undergoing reprogramming indicated that the average amount of target mRNA per cell is within sc-GEM’s linear range of analysis across multiple genes, including somatic genes (a, TFAP2A; b, LUM), pluripotent genes (c, NANOG; d, SOX2; e, ZFP42) and housekeeping gene (f, ACTB), as visualized by comparing the distribution of qPCR threshold cycle number values obtained on single cells to the dynamic range of sc-GEM on bulk RNA standards (horizontal dotted line: mean Ct value +/- 1 standard deviation in single cells [35 fibroblast cells in A-B and 58 human iPS and ES cells in c-f]). (g) Sensitivity of sc-GEM single copy DNA detection was measured at 22 loci that did not contain a CCGG site in 90 single cells. 1972 out of the 1980 single cell PCR reactions produced a specific product, giving a very high estimated successful amplification rate of 99.6%. None of these loci amplified in three microfluidic chambers in the same chip that did not capture any cells, indicating that human contamination is not a major issue in sc-GEM.
Supplementary Figure 3 Correlation between methylation state of a single CpG site and the surrounding region of interest.
Bisulfite sequencing data from ENCODE showed that methylation state of a single CCGG site (dotted line indicate the CCGG site assayed in sc-GEM) is a good proxy for the regional methylation state in representative regions that undergo (a) demethylation and (b) de novo methylation during human cellular reprogramming. Red and green colors indicate methylated and unmethylated CpG sites respectively.
Supplementary Figure 4 Correlation between single cell and bulk gene expression bulk populations.
Single cell ensemble averaged gene expression results during human cellular reprogramming correlate well with bulk gene expression assays results (r = 0.79).
Supplementary Figure 5 Hierarchical clustering of single cell gene expression during human cellular reprogramming.
Hierarchical clustering separated the cells into two distinct clusters. TRA-1-60+ cells in cluster 1 expressed somatic markers (e.g. TFAP2A, TBX3, FOSL1) and early pluripotency markers (e.g. NANOG, SALL4, DNMT3B) whereas cells in cluster 2 expressed early pluripotency markers and late pluripotency markers (e.g. ZIC3, ZFP42, TFCP2L1). TRA-1-60+ cells in cluster #1 and #2 are classified as early pluripotent cells and late pluripotent cells respectively.
Supplementary Figure 6 Comparison of DNA methylation results obtained using SCRAM and bisulfite sequencing.
Single cell ensemble averaged DNA methylation results during human cellular reprogramming is comparable to results obtained using reduced representation bisulfite sequencing from ENCODE (GSE27584).
Supplementary Figure 7 Cell-to-cell correlation matrix of single cell gene expression and single cell DNA methylation.
The data shows the presence of subpopulations of late pluripotent cells within TRA-1-60+ populations at days 16 and 24 and the initiation of DNA methylation changes when cells expressed late pluripotent markers.
Supplementary Figure 8 Boxplot of SALL4 gene expression in d16 and d24 single cells.
Cells were grouped according to the methylation state of the SALL4 promoter measured by sc-GEM. SALL4 gene expression was significantly higher in cells with unmethylated promoter (p = 3.32 x 10-11, Welch Two Sample One-tailed t-test). Rectangle in boxplot spans the first to third quartile. Bold line inside the rectangle shows the median and whiskers above and below the box indicates the maximum and minimum, n is the number of cells in each group.
Supplementary Figure 9 Workflow for tumor sample processing in preparation for sc-GEM.
Lung tumor or non tumor lung samples were surgically removed, dissociated into single cells and stored at -80 °C. Samples were thawed on the day of experiment and stained with propidium iodide. Propidium iodide negative cells were FACS sorted directly onto the Fluidigm C1 chips for sc-GEM experiments.
Supplementary Figure 10 Single cell combined genotype and DNA methylation profiling reveals tumor-specific patterns of DNA methylation.
Hierarchical clustering of single cell DNA methylation profiles from three tumor samples (LUAD1, LUAD2, LUAD3) and a non-tumor lung sample (NTL2) yielded two distinct clusters. The origins of single cells are indicated on the top color bars and their genotypes are indicated on the bottom color bars. Mutation in Exon 21 is a substitution (L858R) while mutation in Exon 19 is a deletion (L747-T751 del).
Supplementary Figure 11 t-SNE analysis of single cell DNA methylation data from tumor and normal tissues.
Samples groups are colored according to a) tissue of origin or b) mutation status.
Supplementary Figure 12 Silhouette analysis of LUAD 1 hierarchical clustering based on gene expression.
Cells are optimally separated into two clusters by silhouette analysis.
Supplementary Figure 13 Comparison between mutation calling with next generation sequencing and Sanger sequencing.
Data is based on one PCR amplicon (EGFR Exon 21) in 60 cells from tumor sample of one patient (A165). A mutant call is made if the mutant read ratio with next generation sequencing exceeds 0.05. High concordance (95%) in mutation calling is observed with these two methods.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–13 and Supplementary Table 2 (PDF 1922 kb)
Primer List
Primer sequences for sc-GEM assay (XLSX 27 kb)
Rights and permissions
About this article

Cite this article
Cheow, L., Courtois, E., Tan, Y. et al. Single-cell multimodal profiling reveals cellular epigenetic heterogeneity. Nat Methods 13, 833–836 (2016). https://doi.org/10.1038/nmeth.3961
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nmeth.3961
This article is cited by
-
Increased blood-based intratumor heterogeneity (bITH) is associated with unfavorable outcomes of immune checkpoint inhibitors plus chemotherapy in non-small cell lung cancer
BMC Medicine (2022)
-
Bisulfite-free epigenomics and genomics of single cells through methylation-sensitive restriction
Communications Biology (2021)
-
Multiplex bioimaging of single-cell spatial profiles for precision cancer diagnostics and therapeutics
npj Precision Oncology (2020)
-
Cardelino: computational integration of somatic clonal substructure and single-cell transcriptomes
Nature Methods (2020)
-
Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy
Journal of Hematology & Oncology (2019)
