
Collection |
Collections
Filters
-
Collection Type
-
-
Focus |
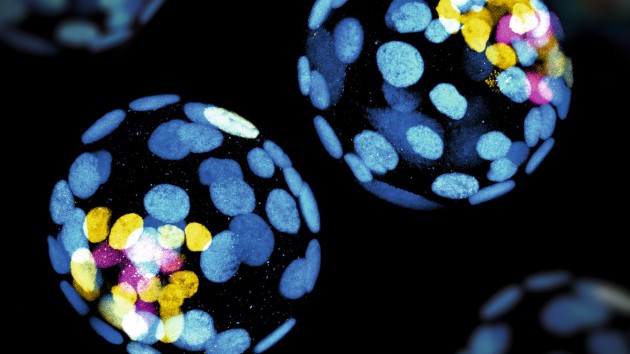 Method of the Year 2023: Methods for modeling development
Method of the Year 2023: Methods for modeling development
Methods for modeling development is our Method of the Year 2023, for the remarkable insights that recent methodological advances have enabled in our understanding of the molecular mechanisms of human embryogenesis.
Image: Berna Sozen, Zernicka-Goetz Lab -
Collection |
 Methods for ecological and evolutionary data analysis
Methods for ecological and evolutionary data analysis
This Collection welcomes primary research articles describing advances in computational and statistical methodology for ecology and evolution.
Image: [M] troyanphoto / stock.adobe.comOpen for submissions -
Collection |
 Nobel Prize in Chemistry 2023
Nobel Prize in Chemistry 2023
The 2023 Nobel Prize in chemistry has been awarded to Moungi G. Bawendi, Louis E. Brus and Alexei I. Ekimov for the discovery and synthesis of quantum dots.
Image: Springer Nature/The Nobel Foundation/Imagesource -
Collection |
 Human BioMolecular Atlas Program
Human BioMolecular Atlas Program
Inaugurated in 2018, the Human BioMolecular Atlas Program (HuBMAP) endeavours to construct comprehensive spatial maps that feature a range of biomolecules such as RNA, proteins, and metabolites in human organs at single-cell resolution.
Image: Heidi Schlehlein -
Focus |
 The future of bioimage analysis
The future of bioimage analysis
Ideas from computer vision have transformed the way image analysis is performed, with ripple effects across microscopy and biological discovery.
Image: Loic Royer and Merlin Lange, Chan Zuckerberg Biohub -
Collection |
 Innovations in Stem Cell Biology 2023
Innovations in Stem Cell Biology 2023
Stem cell models of development, regeneration, and disease are quickly advancing. New technologies and concepts are continuously combined with existing knowledge to create more realistic systems to improve our understanding of these intricate processes. In this collection, we highlight papers published in 2022-2023 across Nature Portfolio journals on topics including embryonic development and stem cells, reproductive biology, synthetic tissues and embryo models, clinical and translational research and tissue stem cells.
Image: Jean-Baptiste Sibarita, Virgile Viasnoff, and Anne Beghin -
Special |
 Human Pangenome Reference
Human Pangenome Reference
The human reference genome is fundamental to basic, translational and clinical research.
Image: Darryl Leja/NHGRI; Sequence map: Adam Novak/UCSC -
Focus |
 Focus on single-cell proteomics
Focus on single-cell proteomics
The emerging field of single-cell proteomics is undergoing a phase of rapid technology development, including advances in mass spectrometry-based methods and single-molecule protein sequencing approaches.
Image: Luis Schachner / Dream by WOMBO / DALL-E by OpenAI -
Collection |
 Extracellular vesicles
Extracellular vesicles
Selected, recent articles from across the Nature Portfolio that document the recent progress in understanding the biology of EV-mediated cell–cell communication and advances in clinical translation of EVs.
Image: Vicky Summersby -
Focus |
 Method of the Year 2022: Long-read sequencing
Method of the Year 2022: Long-read sequencing
Long-read sequencing is our choice for Method of the Year 2022.
Image: Aleksandr Khakimullin / Alamy Stock Photo -
Collection |
Cell cycle
One of the fundamental biological processes in life is the cell cycle leading from DNA replication to cell division. While it has been studied for decades and our knowledge has matured, sophisticated experimental approaches have rejuvenated the field. In addition, cell cycle regulators have emerged as cancer therapy targets. This collection showcases ground-breaking cell cycle papers and reviews, ranging from basic discoveries to clinical applications.
Image: Nicolas Plachta, NCB (2022)
 Best Practices in Method Reporting
Best Practices in Method Reporting
