
Abstract
Type III interferons (IFNs) or IFN-λs regulate a similar set of genes as type I IFNs, but whereas type I IFNs act globally, IFN-λs primarily target mucosal epithelial cells and protect them against the frequent viral attacks that are typical for barrier tissues. IFN-λs thereby help to maintain healthy mucosal surfaces through immune protection, without the significant immune-related pathogenic risk associated with type I IFN responses. Type III IFNs also target the human liver, with dual effects: they induce an antiviral state in hepatocytes, but specific IFN-λ4 action impairs the clearance of hepatitis C virus and could influence inflammatory responses. This constitutes a paradox that has yet to be resolved.
Similar content being viewed by others
Main
Upon infection by viruses, mammals (including humans) react by producing interferons (IFNs), which induce an antiviral state in infected and yet-uninfected cells to block viral replication and spread of the infection. Mammals possess three classes of IFNs: type I (IFN-α/β), type II (IFN-γ) and type III (IFN-λs). The direct antiviral effects of type II IFN are limited, but it has pleiotropic effects on a diverse set of immune cells promoting both adaptive and innate responses. Type I and III IFNs induce a strong antiviral state in responsive cells by initiating a transcriptional program that regulates the expression of several hundred genes. Whereas almost all nucleated cells respond to type I IFN, responses to type III IFNs are restricted to tissues with a high risk of viral exposure and infection, such as those at mucosal surfaces. This allows type III IFNs to selectively induce a strong antiviral state in high-risk tissues at a limited inflammatory cost for the host organism. Here we review the role of IFN-λ as the border guard of the body and discuss the putative roles of IFN-λ4. This paradoxical protein is highly antiviral in vitro but impairs clearance of hepatitis C virus (HCV) in vivo and might influence inflammatory processes in the liver.
Discovery and nomenclature
The type III IFN family was discovered by two teams1,2. The groups chose different naming conventions but subsequently agreed upon the IFN-λ nomenclature3. We use the current nomenclature here and include the now abandoned nomenclature in parentheses. We list protein names, but the names of their encoding genes are generally equivalent, with the exception that Greek letters are replaced by Latin letters (for example, the IFN-λ1 protein is encoded by the IFNL1 gene). In humans, the type III IFN family consists of four members: IFN-λ1 (IL-29), IFN-λ2 (IL28A), IFN-λ3 (IL-28B) and IFN-λ4 (ref. 4). Mice have two functional genes encoding IFN-λ (Ifnl2 and Ifnl3) and two Ifnl1 pseudogenes Ifnl1-P1 and Ifnl1-P2 (Fig. 1). The IFN-λ receptor complex is composed of the specific IFN-λ receptor chain 1 (IFN-λR1 (IL28RA)) and the shared IL-10 receptor chain 2 (IL-10R2 (IL-10Rβ)).

The mouse Ifnl loci are found on chromosome 7 and contain the functional genes Ifnl2 and Ifnl3, and the pseudogenes Ifnl1-P1 and Ifnl1-P2. The duplication of the mouse locus occurred independently from that of the human IFNL locus, so mouse Ifnl2 and Ifln3 cannot be considered true orthologs of human IFNL2 and IFNL3. In humans the IFNL loci are located on the long arm of chromosome 19. In humans IFNL1, IFNL2 and IFNL3 are functional genes, and IFNL4 is a pseudogene in part of the population owing to the SNP rs368234815. Additionally this locus encodes the pseudogenes INFL1P1 and IFNL4P1. SNPs associated with HCV clearance rates are indicated with either their common names or the consequence of the variation in parentheses. The genes IFNL2, IFNL3, IFNL4 and IFNL4P1 were probably generated by a duplication event, whereby the region containing IFNL3 and IFNL4 was duplicated to generate IFNL4P1 and IFNL2, or vice versa. The recombination site localizes between IFNL4 and IFNL4P1. Exons are shown in green; arrows indicate the direction of transcription. Pseudogenes are indicated by gray boxes with the symbol Ψ and are not drawn to scale (except for IFNL4P1). All human pseudogenes contain introns and are thus not the result of retro-transposition.
IFN-λ receptor engagement and signaling
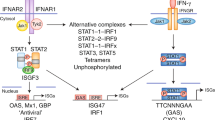
Engagement of the IFN-λ receptor complex by any of the four ligands leads to activation of the receptor-associated tyrosine kinases JAK1 and TYK2, which then phosphorylate specific tyrosines in the intracellular domain of the receptor (Fig. 2). This event creates docking sites for STAT1 and STAT2 signaling molecules, which leads to their recruitment and subsequent phosphorylation1,2,5. The phosphorylated STATs recruit IFN regulatory factor 9 (IRF9), which together form IFN-stimulated gene factor 3 (ISGF3), which enters the nucleus and drives the transcription of IFN-stimulated genes (ISGs). Despite using different receptors, both type I and III IFNs activate ISGF3 (ref. 6) and therefore induce highly similar transcriptional responses6,7,8,9. IFNAR2 (part of the type I IFN receptor complex (Fig. 2)) interacts directly with the IFN-induced protein Usp18, which inhibits the response to IFN-α10 but not to IFN-β or IFN-λ11,12, thereby creating, at least in human cells, a negative feedback loop for IFN-α. In accordance with this, IFN-β and IFN-λ elicit prolonged responses in cell cultures, whereas the response to IFN-α is shorter in duration13,14. However, a different study showed an inhibitory effect of Usp18 on IFN-λ induction of some (but not all) ISGs tested in the mouse system15.

Type I IFNs signal via a heterodimeric receptor composed of the high-affinity chain IFNAR2 and the low-affinity chain IFNAR1. Type III IFNs also signal via a heterodimeric receptor, but it is composed of two different chains: the IFN-λR1 high-affinity chain and IL-10R2 low-affinity chain (which is shared with other cytokines). Both high-affinity chains interact with JAK1, whereas the two low-affinity chains interact with TYK2. Signaling downstream of both receptors results in the assembly of the ISGF3 transcription factor, which coordinates the expression of genes containing an interferon response element (ISRE).
The crystal structure of IFN-λ reveals a four-helix bundle structure typical of class II cytokines, with the closest structural homolog of IFN-λ being interleukin 22 (IL-22)16, but it is not clear whether this reflects a common evolutionary origin or convergence necessitated by the fact that both cytokines utilize IL-10R2. The binding site on the IFN-λR1 receptor chain is well conserved among all four IFN-λs16,17, whereas the binding site on IL-10R2 is poorly defined.
Type III IFN responsive tissues
In mice, the type III IFN response is restricted largely to mucosal epithelial tissues, with the lung epithelium responding to both type I and III IFNs18,19,20 and intestinal epithelial cells responding exclusively to type III IFNs21. Mouse liver shows no expression of IFNλR1 and no responsiveness to IFN-λ in vivo22,23, but liver cells derived from mice require the IFNλR1 receptor for efficient control of viral replication24. In humans, mucosal epithelial tissues as well as the liver respond to type III IFNs25,26. Type III IFN responses in immune cells are still being investigated and are discussed below. Differential splicing of the IFNLR1 gene gives rise to three mRNAs. Variant 1 leads to the expression of the functional receptor; variant 2 lacks a part of the intracellular domain and is nonfunctional; variant 3 encodes only the extracellular part of the receptor27. The product of variant 3 is released from cells and can act as a decoy receptor and potentially downregulate type III IFN responses28, but the physiological role of splice variants 2 and 3 are not yet well established. Interestingly, epigenetic silencing seems to block the induction of IFNLR1 in nonresponsive tissues29, but more work is needed to understand the mechanisms regulating IFNLR1 induction and the physiological role of its splice variants.
Expression of type III IFNs
As would be expected from an antiviral cytokine, and much like the type I IFNs1, type III IFNs can be induced by a wide range of viruses in different cell types30,31,32. Type III IFN can be expressed in a variety of primary human cell types of the hematopoietic lineage33,34,35,36,37, but these cell types also produce type I IFN in abundance. Among nonhematopoietic cells, epithelial cells are potent producers of type III IFNs38,39,40. In mouse models, type III IFNs seem to be the primary type of IFN found in the bronchoalveolar lavage in response to influenza A virus infection41.
The induction of IFNs is mediated by pattern-recognition receptors that recognize the invading virus and initiate a transcriptional response through the transcription factors NF-κB, IRF3 and IRF7 (ref. 42). Early studies on the genes encoding type III IFNs have shown that they have binding sites for the transcription factors NF-κB, IRF3, IRF7 and AP-1 in their promoter regions42,43,44 and can therefore be coexpressed with type I IFNs. Additionally, it was suggested that induction of IFNL1 resembles that of IFNB1, as it seems to be well induced by both IRF3 and IRF7, whereas IFNL2 and IFNL3, similarly to IFNA, are more dependent on IRF7 and seem to have delayed expression kinetics.
It later became clear that the expression of type I and type III IFNs is not regulated by identical mechanisms and could differ among the cell types and stimuli tested. IFNL1, unlike IFNB1, possesses a cluster of distal NF-κB sites that are necessary for maximal IFNL1 transcription and could stimulate IFNL1 gene induction in an IRF-independent manner45. In colon and respiratory epithelial cells, ZEB1 was identified as a selective repressor of IFNL1 transcription, suggesting a key difference between regulation of type I and III IFN expression46,47. Furthermore, induction of type III IFNs can also be initiated via the signaling adaptor MAVS when it is associated with peroxisomes48. The involvement of peroxisomes in the induction of type III IFNs is interesting, as RIG-I–like receptor signaling via MAVS on peroxisomes does not drive the induction of type I IFNs but induces expression of ISGs49. In human hepatocytes infected with HCV or treated with poly(I:C), the induction of IFNL2 and IFNL3 was dependent on IRF3 and IRF7, whereas the induction of IFNL1 was also dependent on NF-κB50. Finally, another group has identified Med23, a subunit of the mediator complex51, as a direct interaction partner for IRF7 (ref. 52). Med23 and IRF7 synergistically increase IFNL transcription but have no effect on IFNB1.
Type I and type III IFNs and the mucosal immune response
The role of type III IFN has been assessed in a number of viral infections, either through the addition of recombinant IFN-λ or in IFN-λR1–deficient mice. The redundant and unique roles of type I and type III IFNs have also been investigated. Not surprisingly, viral tropism is the major determinant of the relative contribution of each IFN type. Gut epithelial cells respond exclusively to type III IFN21, and the type III IFN system mediates control of epitheliotropic viruses, such as rotaviruses, in a nonredundant fashion21. Reoviruses initiate their infection in the gut epithelia but can penetrate the epithelial layer and infect cells in the lamina propria or cause a systemic infection in mice. Type III IFN restricts the initial replication in the gut epithelium and diminishes the shedding of virus through feces, but the type I IFN system is indispensable for the prevention of systemic infection53. Thus, both IFN systems are required in a nonredundant fashion for control of reovirus infection. Similarly, in norovirus infection, type I IFN restricts systemic spread of the virus, but virus control in the gastrointestinal tract is achieved only in the presence of type III IFN signaling54. Surprisingly, type III IFN can eliminate norovirus even in the absence of an adaptive immune system. The protective effect of type III IFN is counteracted by gut commensals55, which explains why wild-type mice with a functional type III IFN system are still susceptible to norovirus. Sterilizing the gut by antibiotic treatment renders mice more resistant to norovirus in a type III IFN–dependent manner55. Many viruses are released and spread from epithelial surfaces; one could thus expect a role for type III IFN in viral transmission, something that should be studied more closely in the future. An early study of the IFN-λR1 knockout mouse revealed a deficiency in the Toll-like receptor 3 (TLR3)- and TLR9-mediated antiviral defense during vaginal infection with herpes simplex virus 2 (HSV-2) but not during systemic viral infection56. Thus, type III IFNs have a role in the vaginal epithelia, but further studies are required in this area. An indirect anti–West Nile Virus effect of type III IFN was reported, in which tight junction integrity of mouse brain microvascular endothelial cells and maintenance of the blood brain barrier required IFN-λR1, thus limiting neuroinvasion57. This effect seems to occur through a noncanonical signaling pathway that requires IFN-λR1 but not STAT1 or protein synthesis.
The compartmentalization of type I and III IFNs is less black and white in the respiratory tract, where there is a degree of redundancy between the two IFN systems18,58. Studies on mice deficient in IFN-λR1, IFNAR1 or both showed that mice lacking both (receptor double-deficient mice) were highly susceptible to respiratory infections, whereas single-deficient mice were largely as resistant as wild-type mice. This indicates a higher degree of redundancy of the two IFN systems in the respiratory tract than in the gastrointestinal tract. Infection of airway epithelial cells in vitro and subsequent measures of the IFN-induced transcriptional signature by microarray showed that the influenza-induced IFN signature was abolished only in receptor double-deficient epithelia9, thus confirming the redundancy observed in vivo. Thus, airway epithelia express receptors for both IFN types, whereas some or all gut epithelial cells seem to express only IFN-λ receptors.
It remains to be seen whether there are niches in the respiratory tract where cells express only IFN-λR1 and where type III IFNs have unique roles, as in the gut. One may speculate that the gut, with its high risk of uncontrolled inflammation owing to the vast burden of microbe-associated molecular patterns derived from commensals, is a compartment where it is desirable to keep local responses 'below the radar' of systemic immunity. In contrast, in the lungs, which are not sterile but certainly have a bacterial burden several orders of magnitude below that of the intestines, the compartmentalization of the two IFN systems is less strict. During influenza virus infection mouse airway epithelia produce higher amounts of type III IFNs than type I IFNs9,41. Therefore, as long as lung epithelial cells are the main IFN producers, the response may be dominated by type III IFNs. However, once immune cells start producing IFN, the response may shift toward being driven by type I IFN. Thus, the division of labor between type III and type I IFNs in the lung might not be as strict as in the gut but seems to follow a similar pattern.
Taken together, observations in the mouse model paint a clear picture of a type III IFN defense system that is active at the borders, defending the mucosal lining against the frequent challenge from viruses. It does so at a substantially lower risk of immune-associated pathology than the type I IFN system would impose, as discussed below. We believe that the lessons learned from the mouse model hold largely true for humans, where type III IFN has an extended role providing protection also against hepatotropic viruses26,59.
Effects of type III IFN on immune cells
Although it is largely accepted that IFN-λR1 expression is more restricted than that of IFNAR1 and IFNAR2, there is still some controversy about which immune cells produce or respond to type III IFN and what functional consequences type III IFN signaling has in immune cells. A consensus is emerging showing that human and mouse plasmacytoid dendritic cells (pDCs), as well as some conventional DC subsets (BDCA3+ in humans and CD8+ in mice), produce type III IFNs35,60. Similarly, only some subsets of myeloid immune cells, such as human monocyte–derived macrophages61, pDCs60,61,62 and some dendritic cell subsets in humans and in mice63, respond directly to type III IFN. For natural killer (NK) cells, reports have come to opposite conclusions about the ability of type III IFNs to influence IFN-γ secretion by NK cells64,65,66. However, adoptive transfer experiments in mice using IFN-λR1–deficient (Ifnlr1−/−) NK cells concluded that direct type III effects on NK cells are required for maximal production of IFN-γ and antitumor activity67. Given the controversial findings, there are essentially three possibilities. One is that the type III IFN effects on NK cells are detected only in combination with other stimuli. A second is that only specific NK cell maturation stages are responsive to type III IFN. Or third, the effects of type III IFN observed are indeed indirect.
Finally, type III IFN can suppresses IL-1 and IL-17 responses as well as neutrophil recruitment in arthritis and other inflammation models68, suggesting a direct effect on neutrophils, which express IFN-λR1. These effects are reminiscent of the antagonism between type I IFN and IL-1 described in tuberculosis69. Effects on T cells and B cells were also reported70,71, but it is still unclear whether these effects are direct or indirect through type III IFN–induced changes in antigen-presenting cells, as described above. Several reports suggest that type III IFN 'favors' IL-12 induction, which indicates a role in promoting type 1 over type 2 immune responses61,63,71, and this effect could be used for allergy treatment63,72 and might be helpful for the induction of strong antiviral responses from type 1 helper T (TH1) cells and CD8 T cells.
Owing to the restricted IFN-λR1 expression by immune cells, the immunomodulatory effects of type III IFNs are limited, which is in contrast to the ubiquitous activity of type I IFN during responses to infection69. The wider range of activity of type I IFN is crucial for the control of systemic viral infection, but the risk of increased disease severity is always looming. Multiple reports describe the deleterious effects of inappropriate type I IFN responses during infection73, including massive induction of proinflammatory cytokines and production of apoptosis-inducing molecules on immune cells during acute infection74 and also in chronic disease75,76,77,78,79, where the source of disease-promoting type I IFN often appears to be pDCs. Furthermore, the blockade of adaptive immune responses by excessive production of type I IFN during chronic infection has been described in detail for T cells80,81 and to a lesser degree for B cells71.
These findings all point in a similar direction: type III IFN is the antiviral weapon of choice when a local mucosal response is sufficient to control the virus and when immune-mediated inflammation is a real risk. Only in severe or systemic infections would it be appropriate to 'pull out the big guns', with a systemic response and strong immune activation, with the associated risk of immune-mediated damage or paralysis of adaptive immunity that may compromise or kill the infected organism (Fig. 3).

(a) When respiratory viruses infect airway epithelia, these cells can establish an antiviral state through the autocrine and paracrine action of type I or type III IFNs. Both types are produced by epithelia, which also express the IFN-λ receptor. In cases where this response is dominated by IFN-λ, the epithelial antiviral state can be established without systemic immune cell activation. (b) Immune cell activation, such as when immune cells are strongly recruited into the infected lung or when they are exposed to virus, can trigger a type I IFN–driven immune response. Because type I IFN has pleiotropic immunomodulatory effects, many cell types are activated, with wide-ranging consequences. Only myeloid immune cells are shown here, but innate and adaptive lymphoid populations are activated in addition. Appropriate type I IFN–driven responses will activate innate and adaptive immune functions and lead to virus elimination. The inflammatory processes involved also have pathogenic potential and can therefore lead to immune-mediated epithelial damage. iMC, inflammatory monocyte; AM, alveolar macrophage.
Type III IFN and coinfection
Type I IFN can exaggerate disease and impair clearance in the case of infection with bacteria such as Listeria monocytogenes and Mycobacterium tuberculosis, most probably due to type I IFN suppression of macrophage activation by IFN-γ69,82,83. In human, macrophages, IFN-γ and TLR activation synergize to activate a series of important antibacterial molecules, and this is antagonized by type I IFN. However, type III IFN acts in a markedly different manner, by increasing macrophage responsiveness to IFN-γ61. The net result is that type III IFN 'favors' the production of TH1 cytokines such as IL-12, as described above. This is interesting, as most comparisons between type I IFN– and type III IFN–mediated gene induction show no or only minor quantitative differences. However, further studies are required in this area.
The difference between the effects of type I and type III IFNs might be important especially in the context of viral-bacterial coinfection, which is seen frequently during influenza virus infections, for example, and can lead to severe disease84,85. Therefore, type III IFN is keeping the borders clear of viral infection without causing widespread immune activation, but it could also be better than type I IFN in steering clear of interfering with the antibacterial immune responses required during polymicrobial exposure. Furthermore, it needs to be explored whether type III IFN interferes with TH17 responses, which is particularly important during coinfections involving viruses and extracellular bacteria.
Why does IFN-λ4 impair clearance of HCV?
HCV causes chronic infection in approximately 75% of people, whereas the remaining individuals manage to clear the infection within the first year86. The host genetic background is important for both spontaneous and treatment-induced clearance of the virus. In 2009, several independent consortia, using rather different HCV-infected cohorts, mapped the major genetic determinant of HCV clearance in response to treatment with IFN-α plus ribavirin to the type III IFN loci87,88,89,90. The same association was found in spontaneous clearance of the virus90,91. However, two different single nucleotide polymorphisms (SNPs) were identified as the best predictor of treatment outcome: rs8099917 (shortened hereafter to 917) best predicted the outcome in Asians and people of European descent, whereas rs12979860 (shortened hereafter to 860) was the best predictor in a US cohort of mixed ethnicity (Fig. 1). Both SNPs were originally described as IL28B-related (i.e., IFNL3-related), and they were surprisingly not in complete linkage disequilibrium (meaning that they are not inherited together), as would be expected if they represented the same underlying causative genetic defect. What was more surprising was that only one of the SNPs identified was located within the encoding regions of known type III IFNs (changing Lys70 in IFN-λ3 to arginine, which had no effect on the function of IFN-λ3)92. These findings led to the speculation that these SNPs represented changes in the IFNL3 promoter region and caused differences in IFN-λ3 expression. However, subsequent studies came to divergent conclusions about the SNPs' influence on IFN-λ3 expression, and this issue is still not fully resolved.
A breakthrough came with the discovery of the IFNL4 gene situated within the region determining HCV clearance. It was found that the SNP rs368234815 was superior to SNP 860 in predicting treatment outcome in individuals of African ancestry. The TT allele of rs368234815 disrupts the open reading frame of IFNL4 and is protective in terms of HCV, whereas the ancestral 'ΔG' allele encodes a functional IFN-λ4 and impairs HCV clearance. This finding suggests that IFN-λ4 is the causative agent of HCV clearance failure, but it does not explain why the SNP 917 is a powerful predictor of the treatment outcome in several other populations. The discovery of a second SNP acting in combination with the ΔG SNP resolved this discrepancy. The SNP rs117648444 (shortened hereafter to 444) represents a nonsynonymous change in the coding region of IFNL4, where a proline residue is replaced by serine, resulting in two versions of IFN-λ4: the fully active IFN-λ4–P70 and a much less active IFN-λ4–S70 (ref. 93). By combining the ΔG and 444 SNPs, one can stratify patients into three groups: (i) those having no IFN-λ4, (ii) those having IFN-λ4–P70 and (iii) those having IFN-λ4–S70. Compared to the single SNPs described previously, this stratification led to a significant improvement in the predictive power of genotyping and reflects the existence of a set of distinct haplotypes in humans (a haplotype is a set of tightly linked alleles that are likely to be inherited together). Before the discovery of the IFNL4 gene, elegant work identified the probable causative haplotype by massive parallel sequencing94. In people of European ancestry, this haplotype is specifically tagged by SNP 917 and encodes the IFN-λ4–P70 variant, whereas the 860 SNP marks several haplotypes encoding both the P70 and S70 variants of IFN-λ4 but not the frameshift mutation. In both cases, the patient group having IFN-λ4–S70 is somewhat neglected, and the apparent discrepancy can be resolved by genotyping and stratification as described above. Notably, this has been done only for one European HCV-infected cohort so far, and its repetition in different cohorts will be important. In particular, the impact of the IFN-λ4–S70 variant might differ among diseases and ethnic backgrounds. In summary, the higher rate of HCV clearance in patients encoding the IFN-λ4–S70 variant than in patients with IFN-λ4–P70 strongly suggests that the activity of the IFN-λ4 protein causes, by yet unknown means, poor HCV clearance.
The effect of IFNL4 genotype is not restricted to IFN-based therapies for HCV; it also extends to direct-acting antiviral-based treatments95,96,97,98,99. Furthermore, IFNL4 genotype also influences the reactivation of cytomegalovirus (CMV) in immune-suppressed patients100,101. The data are clear but paradoxical. IFN-λ4 signals through the same receptor that other members of the type III IFN family do, and its effect is highly similar. Furthermore, IFN-λ4 is antiviral in vitro102. Nevertheless, having a functional IFNL4 gene renders humans less capable of clearing chronic infections such as HCV and CMV. It is likely that IFN-λ4 directly or indirectly influences inflammatory responses and, thereby, viral clearance. Several studies have reported increased liver inflammation and fibrosis103,104,105,106 and increased degranulation activity of lymphocytes107 in HCV patients with the protective IFNL4 genotype (the TT allele, which destroys the open reading frame of IFNL4), suggesting that IFN-λ4 impairs HCV clearance but diminishes liver inflammation and fibrosis. Other studies did not find any association or had directly conflicting data but used smaller cohorts. Interestingly, a study has found that decreased liver inflammation and fibrosis was associated with having a functional IFNL4 gene in a nonalcoholic fatty liver disease cohort, indicating that the effect of IFN-λ4 on inflammation could be independent of viral infection104. However, this finding needs confirmation from other studies. We are currently incapable of offering a mechanistic explanation.
The IFNL4 gene became a liability during human evolution
A report identified IFN-λ4 sequences in most mammalian species, with the notable exception of rodents108. Phylogenetic analyses (Fig. 4) reveal that the IFN-λ4 family constitutes a separate clade in the type III IFN tree. In contrast, IFN-λs 1–3 do not form separate clades but group according to species. This suggests that the ancestor of mammals had an IFNL4-like gene and a second gene that was independently duplicated several times during mammalian evolution, giving rise to IFNL1, IFNL2 and IFNL3. Thus, mouse IFN-λ2 cannot be considered a strict ortholog of human IFN-λ2, etc., similarly to what is seen for IFN-α.

We selected species so that they represented different branches of the mammalian clade. We selected one species of marsupials (gray short-tailed opossum; representing early mammalian evolution) and three representative species from the superorder Laurasiatheria (ferret, pig, hedgehog) as well as mouse and human, which we deal with in this review and represent the two major branches of the superorder Euarchontoglires. For the analysis, we kept the protein names as found in the NCBI database and only added numbers to proteins that had redundant names. The sequences were aligned using MUSCLE (http://www.ebi.ac.uk/Tools/msa/muscle/), and the tree was generated by the Maximum Likelihood method based on the JTT matrix-based model. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site.
It is worth noting that in humans, IFN-λ4 shares only ∼30% identity to IFN-λ3, but despite the low sequence conservation, it has preserved its ability to signal102. This shows that until the appearance of the 'TT-pseudogenizing' allele in humans, IFN-λ4 was under purifying selection to preserve its ability to signal via the IFN-λ receptor complex. This was confirmed by a bioinformatics analysis that found clear purifying selection acting on all nonhuman IFNL4 genes108. The frameshift mutation in human IFNL4 was introduced approximately 55,000 years ago, just before the 'Out of Africa' scenario, and was positively selected almost immediately108. This observation raises several interesting questions. Why did IFNL4 suddenly become a liability? What drove (and still drives) the pseudogenization of IFNL4? Importantly, IFNLR1 exhibits a selection against nonsynonymous substitutions, showing a clear evolutionary pressure in favor of maintaining a functional type III IFN system while specifically eliminating the IFNL4 gene109.
Clinical application of type III IFN
PEGylated IFN-λ1 first entered into clinical trials against HCV, and the initial trial design aimed at replacing PEGylated IFN-α with pegylated IFN-λ1. A successful phase II trial showed that IFN-λ1 was as effective or more so than PEGylated IFN-α and had significantly fewer extrahepatic adverse events59. However, the successful development of several direct-acting antivirals is changing the therapeutic landscape for HCV, and IFN is likely to have a less dominant role in the future. However, several phase III trials are currently under way in which PEGylated IFN-λ1 is combined with direct-acting antivirals (telaprevir, asunaprevir or daclatasvir). Type III IFN could represent a promising future antiviral treatment for some infections with epithelial tropism, as the immunopathological side effects should be minimal. Type III IFN could be useful as a therapy for respiratory infections caused by viruses such as influenza virus or coronaviruses (SARS and MERS)74,110, and for intestinal infection caused by noroviruses or rotaviruses54. Future clinical studies are needed to test the usefulness of type III IFN against these infections and explore the effects of different delivery methods. However, the discovery of IFN-λ4 is also a cautionary lesson, as it appears that in some settings, type III IFN could contribute to increased risk rather than protection from chronic infection.
Concluding remarks
Type III IFN clearly has an important role in protecting the epithelial surfaces from viral infection, but a number of interesting issues remain to be addressed. Given that lung epithelia respond to both type I and type III IFN, what is the distribution of work between these IFN systems in the respiratory epithelia? Humans are frequently challenged with viruses of low or medium pathogenicity, and the particular strength of the type III IFN system might be its ability to deal with these infections without creating severe inflammation or tissue damage. But conclusions drawn from experiments with mice in a highly controlled laboratory environment run the risk of ignoring some of the complexity found in real life. Therefore, in answering future questions, researchers should consider using complex models that allow for viral and bacterial coinfection as well as recurrent infection with several different viruses. Emerging evidence suggests an interaction between type III IFN and the adaptive immune system, but how this occurs is not yet clear. One can imagine direct and indirect effects of type III IFN, through influencing the production of other cytokines, such as IL-12, or via effects on antigen-presenting cells. It will be exciting to follow the development in this area in the coming years. The role of the paradoxical IFN-λ4 in the type III IFN family is currently unclear. There is strong genetic evidence that IFN-λ4 inhibits clearance of chronic viral infections such as HCV and CMV, but at present there is little or no mechanistic understanding of how this occurs.
References
Kotenko, S.V. et al. IFN-λ s mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 4, 69–77 (2003).
Sheppard, P. et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 4, 63–68 (2003). These back-to-back papers reported the discovery of IFN-λs 1–3, describe how they use the IFNλR1 and IL-10R2 receptor chains for signaling and demonstrate their antiviral activity.
Fox, B.A., Sheppard, P.O. & O'Hara, P.J. The role of genomic data in the discovery, annotation and evolutionary interpretation of the interferon-λ family. PLoS ONE 4, e4933 (2009).
Prokunina-Olsson, L. et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat. Genet. 45, 164–171 (2013). This article described the discovery of the IFNL4 gene and demonstrates its association with HCV clearance.
Dumoutier, L. et al. Role of the interleukin (IL)-28 receptor tyrosine residues for antiviral and antiproliferative activity of IL-29/interferon-λ 1: similarities with type I interferon signaling. J. Biol. Chem. 279, 32269–32274 (2004).
Zhou, Z. et al. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. J. Virol. 81, 7749–7758 (2007).
Doyle, S.E. et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 44, 896–906 (2006).
Marcello, T. et al. Interferons α and λ inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 131, 1887–1898 (2006).
Crotta, S. et al. Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza-infected airway epithelia. PLoS Pathog. 9, e1003773 (2013).
Malakhova, O.A. et al. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 25, 2358–2367 (2006).
François-Newton, V. et al. USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon α response. PLoS ONE 6, e22200 (2011).
Makowska, Z., Duong, F.H., Trincucci, G., Tough, D.F. & Heim, M.H. Interferon-β and interferon-λ signaling is not affected by interferon-induced refractoriness to interferon-α in vivo. Hepatology 53, 1154–1163 (2011).
Bolen, C.R., Ding, S., Robek, M.D. & Kleinstein, S.H. Dynamic expression profiling of type I and type III interferon-stimulated hepatocytes reveals a stable hierarchy of gene expression. Hepatology 59, 1262–1272 (2014).
Jilg, N. et al. Kinetic differences in the induction of interferon stimulated genes by interferon-α and interleukin 28B are altered by infection with hepatitis C virus. Hepatology 59, 1250–1261 (2014).
Burkart, C. et al. Usp18 deficient mammary epithelial cells create an antitumour environment driven by hypersensitivity to IFN-λ and elevated secretion of Cxcl10. EMBO Mol. Med. 5, 967–982 (2013).
Gad, H.H. et al. Interferon-λ is functionally an interferon but structurally related to the IL-10 family. J. Biol. Chem. 284, 20869–20875 (2009). Presented the first crystal structure of an IFN-λ, here IFN-λ3, and mapped the IFNλR1 binding site by a mutagenesis approach.
Miknis, Z.J. et al. Crystal structure of human interferon-λ1 in complex with its high-affinity receptor interferon-λR1. J. Mol. Biol. 404, 650–664 (2010).
Mordstein, M. et al. λ interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J. Virol. 84, 5670–5677 (2010).
Sommereyns, C., Paul, S., Staeheli, P. & Michiels, T. IFN-λ (IFN-λ) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 4, e1000017 (2008). Showed that IFN-λs primarily act on epithelial cells and suggested that they evolved to protect epithelial tissues from viral infection.
Pulverer, J.E. et al. Temporal and spatial resolution of type I and III interferon responses in vivo. J. Virol. 84, 8626–8638 (2010).
Pott, J. et al. IFN-λ determines the intestinal epithelial antiviral host defense. Proc. Natl. Acad. Sci. USA 108, 7944–7949 (2011). Found that IFN-λ is required for control of rotavirus infection in mice and showed that type I IFN cannot substitute. This was the first demonstration of a unique role for IFN-λ.
Hermant, P. et al. Human but not mouse hepatocytes respond to interferon-λ in vivo. PLoS ONE 9, e87906 (2014).
Hermant, P. & Michiels, T. Interferon-λ in the context of viral infections: production, response and therapeutic implications. J. Innate Immun. 6, 563–574 (2014).
Anggakusuma et al. Control of hepatitis C virus replication in mouse liver-derived cells by MAVS-dependent production of type I and type III interferons. J. Virol. 89, 3833–3845 (2015).
Meager, A., Visvalingam, K., Dilger, P., Bryan, D. & Wadhwa, M. Biological activity of interleukins-28 and -29: comparison with type I interferons. Cytokine 31, 109–118 (2005).
Muir, A.J. et al. Phase 1b study of PEGylated interferon-λ1 with or without ribavirin in patients with chronic genotype 1 hepatitis C virus infection. Hepatology 52, 822–832 (2010).
Lauber, C. et al. Transcriptome analysis reveals a classical interferon signature induced by IFNλ4 in human primary cells. Genes Immun. doi:10.1038/gene.2015.23 (11 June 2015).
Witte, K. et al. Despite IFN-λ receptor expression, blood immune cells, but not keratinocytes or melanocytes, have an impaired response to type III interferons: implications for therapeutic applications of these cytokines. Genes Immun. 10, 702–714 (2009).
Ding, S., Khoury-Hanold, W., Iwasaki, A. & Robek, M.D. Epigenetic reprogramming of the type III interferon response potentiates antiviral activity and suppresses tumor growth. PLoS Biol. 12, e1001758 (2014).
Durbin, R.K., Kotenko, S.V. & Durbin, J.E. Interferon induction and function at the mucosal surface. Immunol. Rev. 255, 25–39 (2013).
Ank, N. et al. λ interferon (IFN-λ), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J. Virol. 80, 4501–4509 (2006).
Donnelly, R.P. & Kotenko, S.V. Interferon-λ : a new addition to an old family. J. Interferon Cytokine Res. 30, 555–564 (2010).
Coccia, E.M. et al. Viral infection and Toll-like receptor agonists induce a differential expression of type I and λ interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur. J. Immunol. 34, 796–805 (2004).
Osterlund, P. et al. Gene expression and antiviral activity of α/β interferons and interleukin-29 in virus-infected human myeloid dendritic cells. J. Virol. 79, 9608–9617 (2005).
Lauterbach, H. et al. Mouse CD8α+ DCs and human BDCA3+ DCs are major producers of IFN-λ in response to poly IC. J. Exp. Med. 207, 2703–2717 (2010).
Hillyer, P. et al. Expression profiles of human interferon-α and interferon-λ subtypes are ligand- and cell-dependent. Immunol. Cell Biol. 90, 774–783 (2012).
Melchjorsen, J., Siren, J., Julkunen, I., Paludan, S.R. & Matikainen, S. Induction of cytokine expression by herpes simplex virus in human monocyte-derived macrophages and dendritic cells is dependent on virus replication and is counteracted by ICP27 targeting NF-kappaB and IRF-3. J. Gen. Virol. 87, 1099–1108 (2006).
Khaitov, M.R. et al. Respiratory virus induction of α-, β- and λ-interferons in bronchial epithelial cells and peripheral blood mononuclear cells. Allergy 64, 375–386 (2009).
Wang, J. et al. Differentiated human alveolar type II cells secrete antiviral il-29 (IFN-1) in response to influenza A infection. J. Immunol. 182, 1296–1304 (2009).
Ioannidis, I., Ye, F., McNally, B., Willette, M. & Flano, E. Toll-like receptor expression and induction of type I and type III interferons in primary airway epithelial cells. J. Virol. 87, 3261–3270 (2013).
Jewell, N.A. et al. λ interferon is the predominant interferon induced by influenza A virus infection in vivo. J. Virol. 84, 11515–11522 (2010).
Osterlund, P.I., Pietila, T.E., Veckman, V., Kotenko, S.V. & Julkunen, I. IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN-λ ) genes. J. Immunol. 179, 3434–3442 (2007).
Onoguchi, K. et al. Viral infections activate types I and III interferon genes through a common mechanism. J. Biol. Chem. 282, 7576–7581 (2007).
Kotenko, S.V. IFN-λs. Curr. Opin. Immunol. 23, 583–590 (2011).
Thomson, S.J. et al. The role of transposable elements in the regulation of IFN-λ1 gene expression. Proc. Natl. Acad. Sci. USA 106, 11564–11569 (2009).
Siegel, R., Eskdale, J. & Gallagher, G. Regulation of IFN-λ1 promoter activity (IFN-λ1/IL-29) in human airway epithelial cells. J. Immunol. 187, 5636–5644 (2011).
Swider, A., Siegel, R., Eskdale, J. & Gallagher, G. Regulation of interferon λ-1 (IFNL1/IFN-λ1/IL-29) expression in human colon epithelial cells. Cytokine 65, 17–23 (2014).
Odendall, C. et al. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat. Immunol. 15, 717–726 (2014).
Dixit, E. et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 141, 668–681 (2010).
Lee, H.C., Narayanan, S., Park, S.J., Seong, S.Y. & Hahn, Y.S. Transcriptional regulation of IFN-λ genes in hepatitis C virus-infected hepatocytes via IRF-3.IRF-7.NF-κB complex. J. Biol. Chem. 289, 5310–5319 (2014).
Poss, Z.C., Ebmeier, C.C. & Taatjes, D.J. The Mediator complex and transcription regulation. Crit. Rev. Biochem. Mol. Biol. 48, 575–608 (2013).
Griffiths, S.J. et al. A systematic analysis of host factors reveals a Med23-interferon-λ regulatory axis against herpes simplex virus type 1 replication. PLoS Pathog. 9, e1003514 (2013).
Mahlakõiv, T., Hernandez, P., Gronke, K., Diefenbach, A. & Staeheli, P. Leukocyte-derived IFN-α/β and epithelial IFN-λ constitute a compartmentalized mucosal defense system that restricts enteric virus infections. PLoS Pathog. 11, e1004782 (2015).
Nice, T.J. et al. Interferon-λ cures persistent murine norovirus infection in the absence of adaptive immunity. Science 347, 269–273 (2015).
Baldridge, M.T. et al. Commensal microbes and interferon-λ determine persistence of enteric murine norovirus infection. Science 347, 266–269 (2015). Refs. 54 and 55 described that IFN-λ is necessary for a control of norovirus infection in mice. It was shown that recombinant IFN-λ can cure norovirus infection and thus has a significant therapeutic potential. The authors observed that sterilizing the gut by antibiotic treatment cures the persistent virus infection in a manner that requires the IFNλR1 receptor.
Ank, N. et al. An important role for type III interferon (IFN-λ/IL-28) in TLR-induced antiviral activity. J. Immunol. 180, 2474–2485 (2008).
Lazear, H.M. et al. Interferon-λ restricts West Nile virus neuroinvasion by tightening the blood-brain barrier. Sci. Transl. Med. 7, 284ra259 (2015).
Mordstein, M. et al. Interferon-λ contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog. 4, e1000151 (2008).
Muir, A.J. et al. A randomized phase 2b study of peginterferon λ-1a for the treatment of chronic hcv infection. J. Hepatol. 61, 1238–1246 (2014).
Yin, Z. et al. Type III IFNs are produced by and stimulate human plasmacytoid dendritic cells. J. Immunol. 189, 2735–2745 (2012).
Liu, B.S., Janssen, H.L. & Boonstra, A. IL-29 and IFNα differ in their ability to modulate IL-12 production by TLR-activated human macrophages and exhibit differential regulation of the IFNγ receptor expression. Blood 117, 2385–2395 (2011).
Megjugorac, N.J., Gallagher, G.E. & Gallagher, G. Modulation of human plasmacytoid DC function by IFN-λ 1 (IL-29). J. Leukoc. Biol. 86, 1359–1363 (2009).
Koltsida, O. et al. IL-28A (IFN-λ2) modulates lung DC function to promote Th1 immune skewing and suppress allergic airway disease. EMBO Mol. Med. 3, 348–361 (2011).
Dring, M.M. et al. Innate immune genes synergize to predict increased risk of chronic disease in hepatitis C virus infection. Proc. Natl. Acad. Sci. USA 108, 5736–5741 (2011).
Krämer, B. et al. Do λ-IFNs IL28A and IL28B act on human natural killer cells? Proc. Natl. Acad. Sci. USA 108, E519–E520 (2011).
de Groen, R.A. et al. IFN-λ-mediated IL-12 production in macrophages induces IFN-γ production in human NK cells. Eur. J. Immunol. 45, 250–259 (2015).
Souza-Fonseca-Guimaraes, F. et al. NK cells require IL-28R for optimal in vivo activity. Proc. Natl. Acad. Sci. USA 112, E2376–E2384 (2015).
Blazek, K. et al. IFN-λ resolves inflammation via suppression of neutrophil infiltration and IL-1β production. J. Exp. Med. 212, 845–853 (2015).
McNab, F., Mayer-Barber, K., Sher, A., Wack, A. & O'Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 15, 87–103 (2015).
Dai, J., Megjugorac, N.J., Gallagher, G.E., Yu, R.Y. & Gallagher, G. IFN-λ1 (IL-29) inhibits GATA3 expression and suppresses Th2 responses in human naive and memory T cells. Blood 113, 5829–5838 (2009).
Egli, A. et al. IL-28B is a key regulator of B- and T-cell vaccine responses against influenza. PLoS Pathog. 10, e1004556 (2014).
Edwards, M.R. & Johnston, S.L. Interferon-λ as a new approach for treatment of allergic asthma? EMBO Mol. Med. 3, 306–308 (2011).
Davidson, S., Maini, M.K. & Wack, A. Disease-promoting effects of type I interferons in viral, bacterial, and coinfections. J. Interferon Cytokine Res. 35, 252–264 (2015).
Davidson, S., Crotta, S., McCabe, T.M. & Wack, A. Pathogenic potential of interferon alphabeta in acute influenza infection. Nat. Commun. 5, 3864 (2014).
Bosinger, S.E. et al. Global genomic analysis reveals rapid control of a robust innate response in SIV-infected sooty mangabeys. J. Clin. Invest. 119, 3556–3572 (2009).
Jacquelin, B. et al. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J. Clin. Invest. 119, 3544–3555 (2009).
Mandl, J.N. et al. Divergent TLR7 and TLR9 signaling and type I interferon production distinguish pathogenic and nonpathogenic AIDS virus infections. Nat. Med. 14, 1077–1087 (2008).
Micco, L. et al. Differential boosting of innate and adaptive antiviral responses during pegylated-interferon-α therapy of chronic hepatitis B. J. Hepatol. 58, 225–233 (2013).
Penna, A. et al. Peginterferon-α does not improve early peripheral blood HBV-specific T-cell responses in HBeAg-negative chronic hepatitis. J. Hepatol. 56, 1239–1246 (2012).
Teijaro, J.R. et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340, 207–211 (2013).
Wilson, E.B. et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340, 202–207 (2013).
Rayamajhi, M., Humann, J., Penheiter, K., Andreasen, K. & Lenz, L.L. Induction of IFN-γγ enables Listeria monocytogenes to suppress macrophage activation by IFN-γ. J. Exp. Med. 207, 327–337 (2010).
Teles, R.M. et al. Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science 339, 1448–1453 (2013).
Antonelli, L.R. et al. Intranasal Poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J. Clin. Invest. 120, 1674–1682 (2010).
Redford, P.S. et al. Influenza A virus impairs control of Mycobacterium tuberculosis coinfection through a type I interferon receptor-dependent pathway. J. Infect. Dis. 209, 270–274 (2014).
Shepard, C.W., Finelli, L. & Alter, M.J. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 5, 558–567 (2005).
Ge, D. et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 461, 399–401 (2009). This was the first demonstration of a clear genetic association between the IFN-λ loci and the poor treatment outcome for chronic HCV infection.
Suppiah, V. et al. IL28B is associated with response to chronic hepatitis C interferon-α and ribavirin therapy. Nat. Genet. 41, 1100–1104 (2009).
Tanaka, Y. et al. Genome-wide association of IL28B with response to PEGylated interferon-α and ribavirin therapy for chronic hepatitis C. Nat. Genet. 41, 1105–1109 (2009).
Rauch, A. et al. Genetic variation in IL28B is associated with chronic hepatitis C and treatment failure: a genome-wide association study. Gastroenterology 138, 1338–1345 (2010).
Thomas, D.L. et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 461, 798–801 (2009).
Urban, T.J. et al. IL28B genotype is associated with differential expression of intrahepatic interferon-stimulated genes in patients with chronic hepatitis C. Hepatology 52, 1888–1896 (2010).
Terczyn´ska-Dyla, E. et al. Reduced IFN-λ4 activity is associated with improved HCV clearance and reduced expression of interferon-stimulated genes. Nat. Commun. 5, 5699 (2014). This article provided the first direct connection between the activity of the IFN-λ4 protein, the high expression of interferon stimulated genes in the liver and poor clearance rate of chronic HCV.
Smith, K.R. et al. Identification of improved IL28B SNPs and haplotypes for prediction of drug response in treatment of hepatitis C using massively parallel sequencing in a cross-sectional European cohort. Genome Med. 3, 57 (2011).
Ahlenstiel, G., Booth, D.R. & George, J. Will IL28B polymorphisms remain relevant to direct-acting antiviral treatment paradigms? Antivir. Ther. 17, 1163–1170 (2012).
Holmes, J.A., Desmond, P.V. & Thompson, A.J. Does IL28B genotyping still have a role in the era of direct-acting antiviral therapy for chronic hepatitis C infection? J. Viral Hepat. 19, 677–684 (2012).
Meissner, E.G. et al. IFNL4-ΔG genotype is associated with slower viral clearance in hepatitis C, genotype-1 patients treated with sofosbuvir and ribavirin. J. Infect. Dis. 209, 1700–1704 (2014).
Muir, A.J. IL28B in the era of direct-acting antivirals for hepatitis C. J. Clin. Gastroenterol. 47, 222–227 (2013).
Zeuzem, S. et al. Faldaprevir and deleobuvir for HCV genotype 1 infection. N. Engl. J. Med. 369, 630–639 (2013).
Bibert, S. et al. The IFNL3/4 ΔG variant increases susceptibility to cytomegalovirus retinitis among HIV-infected patients. AIDS 28, 1885–1889 (2014).
Manuel, O. et al. Influence of IFNL3/4 polymorphisms on the incidence of cytomegalovirus infection after solid-organ transplantation. J. Infect. Dis. 211, 906–914 (2015).
Hamming, O.J. et al. Interferon-λ4 signals via the IFN-λ receptor to regulate antiviral activity against HCV and coronaviruses. EMBO J. 32, 3055–3065 (2013).
Bochud, P.Y. et al. IL28B alleles associated with poor hepatitis C virus (HCV) clearance protect against inflammation and fibrosis in patients infected with non-1 HCV genotypes. Hepatology 55, 384–394 (2012).
Eslam, M. et al. Interferon-λ rs12979860 genotype and liver fibrosis in viral and non-viral chronic liver disease. Nat. Commun. 6, 6422 (2015).
Petta, S. et al. IL28B and PNPLA3 polymorphisms affect histological liver damage in patients with non-alcoholic fatty liver disease. J. Hepatol. 56, 1356–1362 (2012).
Noureddin, M. et al. Association of IL28B genotype with fibrosis progression and clinical outcomes in patients with chronic hepatitis C: a longitudinal analysis. Hepatology 58, 1548–1557 (2013).
Jouvin-Marche, E. et al. Lymphocytes degranulation in liver in hepatitis C virus carriers is associated with IFNL4 polymorphisms and ALT levels. J. Infect. Dis. 209, 1907–1915 (2014).
Key, F.M. et al. Selection on a variant associated with improved viral clearance drives local, adaptive pseudogenization of interferon-λ4 (IFNL4). PLoS Genet. 10, e1004681 (2014).
Manry, J. et al. Evolutionary genetic dissection of human interferons. J. Exp. Med. 208, 2747–2759 (2011).
Kindler, E. et al. Efficient replication of the novel human betacoronavirus EMC on primary human epithelium highlights its zoonotic potential. MBio 4, e00611–e00612 (2013).
Acknowledgements
We thank L. Heilesen, H.H. Gad and S. Davidson for critical reading of this manuscript and help with figures.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Wack, A., Terczyńska-Dyla, E. & Hartmann, R. Guarding the frontiers: the biology of type III interferons. Nat Immunol 16, 802–809 (2015). https://doi.org/10.1038/ni.3212
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ni.3212
This article is cited by
-
TRIM26 positively affects hepatitis B virus replication by inhibiting proteasome-dependent degradation of viral core protein
Scientific Reports (2023)
-
Severity of neonatal influenza infection is driven by type I interferon and oxidative stress
Mucosal Immunology (2022)
-
The role of IFNL4 in liver inflammation and progression of fibrosis
Genes & Immunity (2022)
-
The Priming Potential of Interferon Lambda-1 for Antiviral Defense in the Oral Mucosa
Inflammation (2022)
-
IFN-λ4 genetic variants influence clinical malaria episodes in a cohort of Kenyan children
Malaria Journal (2021)
