
Abstract
The kinase mTOR has emerged as an important regulator of the differentiation of helper T cells. Here we demonstrate that differentiation into the TH1 and TH17 subsets of helper T cells was selectively regulated by signaling from mTOR complex 1 (mTORC1) that was dependent on the small GTPase Rheb. Rheb-deficient T cells failed to generate TH1 and TH17 responses in vitro and in vivo and did not induce classical experimental autoimmune encephalomyelitis (EAE). However, they retained their ability to become TH2 cells. Alternatively, when mTORC2 signaling was deleted from T cells, they failed to generate TH2 cells in vitro and in vivo but preserved their ability to become TH1 and TH17 cells. Our data identify mechanisms by which two distinct signaling pathways downstream of mTOR regulate helper cell fate in different ways. These findings define a previously unknown paradigm that links T cell differentiation with selective metabolic signaling pathways.
Similar content being viewed by others


Main
The kinase mTOR is an evolutionarily conserved member of the phosphatidylinositol-3-OH kinase (PI(3)K)-related kinase family that has a central role in the regulation of metabolism, protein synthesis, energy balance, proliferation and survival1. This kinase senses environmental cues such as amino acids, insulin and growth factors and then integrates such signals to regulate cellular metabolism2. It forms the core of two distinct signaling complexes whose activation is regulated differently3,4. The complex mTORC1 contains the scaffolding protein Raptor, as well as the subunits mLST8, PRAS40 and Deptor5. Activation of mTORC1 is achieved through signaling by the kinases PI(3)K, PDK1 and Akt. This complex promotes phosphorylation of the translational regulators S6K1 and 4E-BP1 and is believed to have a central role in regulating cellular growth and proliferation by modulating metabolism6. The second mTOR signaling complex, mTORC2, consists of mLST8, the scaffolding protein Rictor and the subunits mSIN1 and Protor5. Rictor is a critical adaptor protein for mTORC2, and Rictor-deficient mice and mouse embryonic fibroblasts have been shown to lack mTORC2 activity4. The activity of mTORC2 can be measured by phosphorylation of the hydrophobic motif of Akt at Ser473 and other kinases of the AGC (protein kinases A, G and C) family (SGK1 and PKC-α)7. The exact upstream activators of mTORC2 signaling have yet to be fully elucidated. Despite ongoing studies aimed at delineating both upstream activators and downstream effectors of the two respective signaling pathways, the distinct physiological consequences mediated by mTORC1 and mTORC2 have yet to be defined in biological systems.
A central role for mTOR in sensing the immune microenvironment and dictating immune function and differentiation has begun to emerge8. CD4+ T cells that lack mTOR fail to differentiate into effector cells under appropriate skewing conditions9. Instead, after activation, mTOR-deficient T cells become Foxp3+ regulatory cells. This inability to differentiate into effector cells in the absence of mTOR is associated with less activation of the transcription factors STAT4, STAT6 and STAT3 in response to the skewing cytokines interleukin 12 (IL-12), IL-4 and IL-6, respectively. Moreover, mTOR controls the development of CD8+ memory T cells in part by regulating expression of the transcription factors T-bet and eomesodermin10,11. In addition, the PI(3)K-mTOR axis regulates lymphocyte trafficking12.
The small GTPase Rheb is isolated from the central nervous system and was originally thought to be a mediator of the GTPase Ras–mitogen-activated protein kinase pathway13,14. The mammalian Rheb family consists of two proteins, Rheb1 and Rheb2; both are expressed in T cells, but their function has yet to be elucidated13,15. Rheb is a crucial regulator of mTORC1 signaling16. The tuberous sclerosis complex (TSC), composed of TSC1 and TSC2, functions as a GTPase-activating protein for Rheb. When TSC is inhibited, the active, GTP-bound form of Rheb interacts with mTORC1 to stimulate its activity15.
Despite emerging appreciation for the central role of mTOR in regulating the differentiation of effector T cells, memory T cells and regulatory T cells (Treg cells), the downstream signaling pathways involved in regulating these processes have yet to be elucidated. By selectively knocking out Rheb in T cells, we demonstrate here that the differentiation of both T helper type 1 (TH1) and IL-17-producing helper T (TH17) effector cells required mTORC1 signaling, whereas TH2 differentiation was preserved. Mice with T cell–specific deletion of Rheb did not mount TH1 or TH17 responses in vivo and were resistant to the development of classical experimental autoimmune encephalomyelitis (EAE). As T cells lacking mTORC1 activity were still able to become TH2 cells, we further investigated the role of mTORC2 signaling in TH2 development. After deleting mTORC2 signaling in T cells, we found that they were unable to become TH2 cells but maintained their ability to differentiate into TH1 cells and TH17 cells. Our findings define distinct downstream signaling pathways as the mechanisms by which mTOR regulates the differentiation of helper T cells.
Results
Rheb regulates mTORC1 activity in T cells
We hypothesized that genetic deletion of Rheb in T cells would inhibit mTORC1 activation. We bred mice with loxP-flanked Rheb alleles with mice expressing Cre recombinase from the Cd4 promoter and enhancer regions, which led to deletion of Rheb in T cells in the 'T-Rheb−/−' progeny (Supplementary Fig. 1). We stimulated CD4+ T cells from wild-type and T-Rheb−/− mice with antibody to CD3 (anti-CD3) and anti-CD28 and assessed the activity of mTORC1 and mTORC2 biochemically. After stimulation, wild-type T cells showed considerable phosphorylation of S6K1, indicative of mTORC1 activity (Fig. 1a,b). In contrast, S6K1 phosphorylation was almost completely absent in the activated T-Rheb−/− T cells. Phosphorylation of Akt at Thr308 (which is PDK1 dependent and upstream of mTORC1) remained intact in the T-Rheb−/− T cells (Fig. 1a,b). Likewise, phosphorylation of Akt at Ser473 (which is downstream of mTORC2) remained intact.

(a) Immunoblot analysis of mTOR activation, assessed as phosphorylation (p-) of S6K1 and Akt, in lysates of wild-type (WT) and T-Rheb−/− CD4+ T cells stimulated for 1 h or 3 h with anti-CD3 and anti-CD28 (TCR + costim) in serum-free medium. (b) Densitometry analysis of the activity of mTORC1 (p-S6K1), mTORC2 (p-Akt(Ser473)) and PI(3)K (p-Akt(Thr308)) in the cells in a (n = 4 mice per group). NS, not significant. *P < 0.05, **P < 0.01 and ***P < 0.001 (Student's t-test). (c) Flow cytometry of splenocytes from wild-type and T-Rheb−/− mice. Plots of CD3 and B220 (top) are gated on lymphocyte forward and side scatter; plots of CD4 and CD8 (middle) are gated on CD3+ cells; plots of Foxp3 and CD25 Treg cells (bottom) are gated CD3+CD4+ cells. (d) CFSE dilution in wild-type and T-Rheb−/− CD4+ T cells stimulated for 48 h (Day 2) or 96 h (Day 4) with anti-CD3 and irradiated autologous antigen-presenting cells (APCs). (e) IL-2 production in wild-type and T-Rheb−/− CD4+ T cells stimulated overnight with anti-CD3 and anti-CD28. Numbers in quadrants or adjacent to outlined areas (c,d) indicate percent cells in each. Data are representative of at least three independent experiments (c–e) or four experiments (a,b; error bars (b,e), s.e.m.).
Next we sought to determine the functional consequences of deleting Rheb in T cells. Wild-type and T-Rheb−/− mice had a similar frequency of CD8+ T cells, CD4+ T cells and B cells (Fig. 1c and Supplementary Fig. 2). In addition, despite the lack of mTORC1 activity, we did not see any difference between wild-type and T-Rheb−/− mice in the development of Foxp3+ T cells. Similar to T cells lacking mTOR9, T cells from the T-Rheb−/− mice did not demonstrate a block in proliferation but instead proliferated more slowly than their wild-type counterparts (Fig. 1d). Likewise, IL-2 production was similar in wild-type and T-Rheb−/− T cells (Fig. 1e), which indicated that T cell antigen receptor (TCR)-induced activation and subsequent expression of IL-2 was not affected by the absence of Rheb. In some systems, mTOR signaling can affect apoptosis. After in vitro activation, there was no evidence of greater apoptosis in T cells from T-Rheb−/− mice, as assessed by staining with annexin V and 7-amino-actinomycin D (Supplementary Fig. 3). Thus, deleting Rheb in T cells specifically eliminates mTORC1 activity while preserving mTORC2 activity. Furthermore, elimination of Rheb in T cells still permits TCR-induced cytokine expression and proliferation.
TH1 and TH17 differentiation require mTORC1
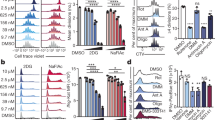
Given that mTOR-deficient T cells fail to differentiate into TH1, TH2 or TH17 effector cells, we wanted to determine the effect of Rheb deletion on T cell differentiation. There was no difference between wild-type and T-Rheb−/− littermates in their naive and memory T cell compartments, as assayed with freshly isolated lymphocytes (Supplementary Fig. 4). Similar to T cells lacking mTOR, T-Rheb−/− T cells failed to differentiate into TH1 cells under the appropriate skewing conditions in vitro (Fig. 2a). T-Rheb−/− T cells also failed to differentiate into TH17 cells (Fig. 2b). These observations identify mTORC1 signaling as the downstream mediator of mTOR-dependent TH1 and TH17 differentiation. In contrast to mTOR-deficient T cells, T-Rheb−/− T cells were able to differentiate into TH2 cells (Fig. 2c). Therefore, inhibition of mTORC1 activity via deletion of Rheb selectively inhibited TH1 and TH17 differentiation but preserved TH2 differentiation.

(a) IFN-γ production in wild-type and T-Rheb−/− CD4+ T cells stimulated with anti-CD3 and APCs in TH1- or TH2-skewing conditions (horizontal axis), followed by 5 d of population expansion with IL-2 and restimulation in vitro with anti-CD3 and anti-CD28. (b) IL-17 production in wild-type and T-Rheb−/− CD4+ T cells stimulated with anti-CD3 and APCs in TH1- or TH17-skewing conditions (left margin), followed by 5 d of population expansion and restimulation with anti-CD3 and APCs in the presence of a protein-transport inhibitor. Numbers in outlined areas indicate percent IL-17+CD4+ cells. (c) IL-4 production in wild-type and T-Rheb−/− CD4+ T cells treated as in a. (d) CFSE dilution and cytokine production of wild-type and T-Rheb−/− CD4+ T cells labeled and skewed as in a and restimulated with anti-CD3 and anti-CD28 in the presence of a protein-transport inhibitor. Numbers in quadrants indicate percent cells in each. Data are representative of at least three independent experiments (error bars (a,c), s.e.m.).
To determine if the defect in TH1 skewing was linked to the proliferative defect in Rheb-deficient T cells, we labeled wild-type and Rheb-deficient T cells with the cytosolic dye CFSE, cultured them in TH1- or TH2-skewing conditions and assessed their cytokine production after rechallenge. Rheb-deficient T cells did proliferate in vitro, although at lower rates. Furthermore, Rheb-deficient T cells that divided several times under TH1-skewing conditions still did not make interferon-γ (IFN-γ) in amounts similar to those of wild-type cells (Fig. 2d). Furthermore, the slightly greater IL-4 production observed did not directly correlate with the number of cell divisions. Thus, the inability of Rheb-deficient T cells to differentiate into TH1 cells was not secondary to their lower rate of proliferation.
Next we sought to confirm in vivo the results reported above. Infection with vaccinia virus leads to a robust TH1 response. We obtained T cells from wild-type and T-Rheb−/− mice crossed onto the OT-II TCR–transgenic background specific for ovalbumin (OVA) and adoptively transferred the cells into wild-type C57BL/6 mice17. We infected the recipient mice with vaccinia virus expressing ovalbumin and evaluated clonotypic T cells 5 d later for expression of IFN-γ and IL-4. As expected, wild-type OT-II cells differentiated into TH1 cells with robust production of IFN-γ, IL-2 and tumor necrosis factor but little IL-4 (Fig. 3a,b). In contrast, T-Rheb−/− clonotypic T cells failed to robustly produce IFN-γ but produced substantial IL-4. To assess the role of proliferation in this in vivo system, we labeled OT-II cells with CFSE. Cells transferred into host mice not infected with virus failed to proliferate or produce cytokines after rechallenge, whereas wild-type T cells transferred into immunized mice proliferated in vivo and produced IFN-γ (Fig. 3c). However, Rheb-deficient T cells, even those undergoing multiple rounds of proliferation, failed to produce IFN-γ. The addition of an IL-4-neutralizing antibody to the culture conditions failed to restore IFN-γ production (data not shown). These data demonstrate that selective deletion of mTORC1 signaling leads to enhanced TH2 differentiation. Thus, selective inactivation of mTORC1 in T cells did not lead to T cell tolerance but instead promoted differentiation down an alternative effector pathway.

(a) Cytokine production in C57BL/6 (Thy-1.2+) mice immunized with vaccinia virus expressing ovalbumin and given adoptive transfer of 1 × 106 wild-type or T-Rheb−/− CD4+ OT-II (Thy-1.1+) T cells; splenocytes obtained 4 d later were rechallenged overnight in vitro with OVA peptide in the presence of a protein-transport inhibitor. Plots are gated on Thy-1.1+ cells. TNF, tumor necrosis factor. (b) Cytokine production by cells from mice treated as in a. Each symbol represents an individual mouse (n = 3); small horizontal lines indicate the mean. P < 0.01 for IFN-γ or 0.05 for IL-4 (Student's t-test). (c) Cytokine production in C57BL/6 (Thy-1.2+) mice given mock immunization or immunized as in a and given adoptive transfer as in a, except with CFSE-labeled cells. (d) Cytokine production from Peyer's patch lymphocytes (PPLs) isolated from wild-type and T-Rheb−/− mice (n = 6) and stimulated with PMA and ionomycin. *P < 0.01 (Student's t-test). Numbers in quadrants (a,c) or outlined areas (d) indicate percent cells in each. Data are representative of at least three independent experiments.
We also assessed the ability of Rheb-deficient T cells to differentiate into TH17 cells in vivo. TH17 cells develop in vivo in the gut in response to pathogens. After brief stimulation with the phorbol ester PMA and ionomycin in vitro, there were considerably fewer in vivo–differentiated CD3+CD4+ TH17 cells in the Peyer's patches of T-Rheb−/− mice than in those of wild-type mice (Fig. 3d). Overall, these data show that TH1 and TH17 differentiation are specifically regulated by mTORC1 signaling in vivo and in vitro but TH2 differentiation is not.
T-Rheb−/− mice are resistant to EAE
Because both TH1 and TH17 cells are linked to the promotion of autoimmune diseases and IFN-γ and IL-17 participate in promoting the pathology seen in EAE, we determined the susceptibility of the T-Rheb−/− mice to this experimental model18. After immunization with myelin oligodendrocyte glycoprotein (MOG) peptide of amino acids 35–55, wild-type mice developed clinical EAE (Fig. 4a), whereas T-Rheb−/− mice had lower clinical scores, associated with less leukocytic infiltration in the spinal cord and less population expansion of TH1 and TH17 cells (Fig. 4b,c). However, nearly 60% (on average) of MOG-immunized T-Rheb−/− mice developed 'nonclassical EAE' (Supplementary Video 1), which is characteristic of TH2 responses to MOG (Fig. 4d). MOG-specific TH2 cells or IFN-γ-deficient cells can induce a neurological disorder characterized by ataxia (not paralysis) and infiltration of the cerebellum with cells of the immune response19. T-Rheb−/− mice had more leukocytic infiltration, including CD4+ cells in the cerebellum (Fig. 4e), and T cells isolated from these mice produced more IL-4 and IL-5 in response to MOG peptide in vitro (data not shown). Thus, mTORC1-deficient T cells were neither 'immunodeficient' nor unable to traffick to the CNS. Therefore, selective deletion of mTORC1 signaling leads to enhanced TH2 differentiation in vivo, even under strongly TH1- and/or TH17-promoting conditions.

(a) Disease progression in wild-type and T-Rheb−/− mice (n = 7) immunized with MOG and complete Freund's adjuvant to induce EAE. *P < 0.01 (one-way analysis of variance). (b) Immunohistochemistry of CD45 and CD4 in sections of the central nervous system from wild-type mice, frozen at the height of disease. (c) Cytokine production in samples from the central nervous system, allowed to 'rest' for 16 h in unsupplemented medium and stimulated with PMA and ionomycin in the presence of a protein-transport inhibitor (n = 4 mice). Plots (left) are gated by CD4 expression and side scatter and numbers in quadrants indicate percent cells in each; each symbol (right) represents an individual mouse. P < 0.05 for IFN-γ or 0.01 for IL-17 (Student's t-test). (d) Incidence of classical or nonclassical EAE in mice immunized with MOG and complete Freund's adjuvant. (e) Immunohistochemistry of CD45 and CD4 in the cerebellum, assessed as in b. Original magnification (b,e), ×20. Data are representative of three independent experiments (error bars (a,d), s.e.m.).
TH2 differentiation requires mTORC2 signaling
We further bred mice with loxP-flanked Rictor alleles with mice expressing Cre recombinase from the Cd4 promoter and enhancer regions (producing 'T-Rictor−/−' progeny) to determine the effect of selectively eliminating mTORC2 in T cells20 (Supplementary Fig. 5). After stimulation with anti-CD3 and anti-CD28, T-Rictor−/− T cells had less mTORC2 activity, as determined by phosphorylation of the Akt hydrophobic motif (Ser473). The activity of mTORC1 in these cells was normal, as determined by phosphorylation of S6K1 (Fig. 5a,b). In agreement with observations obtained with Rictor-deficient mice7, proximal (PI(3)K-PDK1) signaling, assessed as phosphorylation of Akt at Thr308, was intact and consistently greater. T-Rictor−/− CD4+ T cells developed normally (with a slightly lower frequency of peripheral CD8+ cells; Supplementary Fig. 6) and produced amounts of IL-2 equivalent to those produced by wild-type T cells (Supplementary Fig. 7), which indicated that TCR-induced signaling was intact. T-Rictor−/− T cells proliferated more slowly after stimulation but had a less severe defect than T-Rheb−/− T cells had (Supplementary Fig. 7). In addition, T-Rictor−/− mice did not have more activation-induced apoptosis (Supplementary Fig. 3) and had normal proportions of CD4+ T cells in the naive compartment (Supplementary Fig. 8).

(a) Immunoblot analysis of mTOR activation (assessed as in Fig.1a) in lysates of wild-type and T-Rictor−/− CD4+ T cells stimulated for 1 h or 3 h with anti-CD3 and anti-CD28 in serum-free medium. (b) Densitometry analysis of the activity of mTORC1, mTORC2 and PI(3)K (assessed as in 1b; n = 4 mice). *P < 0.05 (Student's t-test). (c) IFN-γ production in wild-type, T-Rheb−/− and T-Rictor−/− CD4+ T cells stimulated overnight with anti-CD3 and APCs in TH1- or TH2-skewing conditions, followed by population expansion for 5 d with IL-2 and restimulation in vitro with anti-CD3 and anti-CD28. (d) IL-17 production by wild-type and T-Rictor−/− CD4+ T cells stimulated with anti-CD3 and APCs in TH1- or TH17-skewing conditions, followed by population expansion for 5 d and restimulation with anti-CD3 and APCs in the presence of a protein-transport inhibitor. Numbers in outlined areas indicate percent IL-17+CD4+ cells. (e) IL-4 production in cells treated as in c. (f) Cytokine production by splenocytes (treated as in Fig. 3a) from C57BL/6 (Thy-1.2+) mice immunized with vaccinia virus expressing ovalbumin and given adoptive transfer of 1 × 106 wild-type, T-Rheb−/− or T-Rictor−/− CD4+ OT-II (Thy-1.1+) T cells. Each symbol represents an individual mouse; small horizontal lines indicate the mean. *P < 0.01 (Student's t-test). (g) IL-4 production in CD4+ T cells from wild-type, T-Rheb−/− and T-Rictor−/− mice immunized with alum (Naive) or ovalbumin plus alum (OVA primed) and boosted after 14 d; splenocytes were stimulated 48 h with OVA protein (100 μg/ml) with protein-transport inhibitor present for the final 8 h. Numbers in outlined areas indicate percent IL-4+CD4+ cells. (h) OVA-specific serum immunoglobulin G1 (IgG1) antibody titers in the mice in g. Data are representative of four (a,b) or two (f) experiments or at least three independent experiments (c–e,g,h; error bars (b,c,e,h), s.e.m.).
T-Rictor−/− T cells were able to develop into TH1 and TH17 cells under the appropriate skewing conditions in vitro (Fig. 5c,d) but failed to differentiate down the TH2 pathway (Fig. 5e). To exclude the possibility that the presence of previously activated cells may have altered skewing conditions in our system, we sorted naive (CD62LhiCD44lo) CD4+ cells from wild-type, T-Rheb−/− and T-Rictor−/− mice and skewed them with plate-bound anti-CD3 and soluble anti-CD28. As seen with bulk CD4+ T cells, Rheb-deficient cells failed to differentiate into TH1 or TH17 cells, and Rictor-deficient cells failed to differentiate into TH2 cells (Supplementary Fig. 9). TCR signaling strength can affect TH1 and TH2 responses21. Specifically, low concentrations of peptide can impair IFN-γ production and create a bias toward the TH2 response. To assess the effect of strength of peptide signaling on the differentiation of Rheb-deficient and Rictor-deficient T cells, we stimulated wild-type, Rheb-deficient and Rictor-deficient OT-II T cells under conditions of widely varying doses of OVA peptide as described before22. However, even at a dose of 100 μM OVA peptide, there was no evidence of more IFN-γ production from Rheb-deficient T cells (Supplementary Fig. 10). In contrast, Rictor-deficient T cells produced TH1 cytokines even at exceedingly low doses of peptide.
Consistent with the in vitro data, T-Rictor−/− T cells had robust production of IFN-γ in response to infection with vaccinia virus (Fig. 5f). T-Rictor−/− mice had normal susceptibility to EAE (Supplementary Fig. 11) associated with numbers of IFN-γ- and IL-17-producing cells in response to MOG peptide equivalent to those in wild-type mice. To assess TH2 responses in vivo, we primed and boosted wild-type (C57BL/6), T-Rheb−/− and T-Rictor−/− mice with OVA in alum as an adjuvant to elicit a TH2 response. T-Rictor−/− mice had less induction of IL-4-secreting OVA-specific T cells than did wild-type or T-Rheb−/− mice (Fig. 5g) and failed to generate antigen-specific immunoglobulin G1 antibodies (which are dependent on TH2 cell help; Fig. 5h). Thus, mTORC2 regulates the ability of helper T cells to differentiate into TH2 cells.
Distinct STAT activation by mTORC1 and mTORC2
Next we determined if we could reproduce the results reported above in wild-type primary T cells by pharmacological inhibition of mTOR activity. Biochemical analysis of T cells showed that rapamycin inhibited mTORC2 activity23 (Fig. 6a). However, very low doses of rapamycin (100–500 pM) selectively inhibit mTORC1 in T cells. Rapamycin at such a very low dose specifically inhibited TH1 differentiation by naive wild-type T cells activated under either TH1- or TH2-skewing conditions (Fig. 6b), consistent with the findings obtained with Rheb-deficient T cells.

(a) Immunoblot analysis of mTOR activation (assessed as in Fig. 1a) in lysates of wild-type, naive T cells stimulated for 8 h with anti-CD3 and anti-CD28 plus various doses of rapamycin (above lanes) in serum-containing medium. (b) Cytokine production by wild-type 5C.C7 T cells stimulated for 48 h with pigeon cytochrome c peptide in TH1- or TH2-skewing conditions in the presence (VLD rapamycin) or absence (Untreated) of a very low dose of rapamycin (100 pM) and washed, followed by population expansion for 5 d in fresh drug and IL-2, then restimulation with anti-CD3 and anti-CD28. Numbers in quadrants indicate percent IL-4+IFN-γ− cells (top left) or IL-4−IFN-γ+ cells (bottom right). (c) Immunoblot analysis of STAT phosphorylation in lysates of freshly isolated wild-type, T-Rheb−/− and T-Rictor−/− CD4+ T cells resuspended for 1 h in serum-free medium and stimulated for 30 min with IL-12, IL-4 or IL-6. (d) SOCS3 and SOCS5 mRNA in wild-type, T-Rheb−/− and T-Rictor−/− CD4+ T cells left unstimulated (Unstim) or activated overnight with anti-CD3 and anti-CD28 (Activated); results are normalized to those of 18s rRNA and are presented relative to those of wild-type unstimulated cells (for SOCS3) or stimulated cells (for SOCS5). (e) Immunoblot analysis of SOCS3 and SOCS5 in cells treated as in d. (f) Kinetic analysis of SOCS3 and SOCS5 in lysates of wild-type, T-Rheb−/− and T-Rictor−/− CD4+ T cells stimulated for 48, 72 or 96 h with anti-CD3 and anti-CD28; actin serves as a loading control. (g) IFN-γ production in wild-type, T-Rheb−/− and T-Rictor−/− CD4+ T cells transfected by nucleofection with control siRNA (siCtrl) or siRNA specific for SOCS3 (siSOCS3) or SOCS5 (siSOCS5; data not shown), allowed to 'rest' for 16 h and then stimulated for 48 h in TH1- or TH2-skewing conditions, followed by population expansion for 48 h in IL-2 and restimulation with anti-CD3 and anti-CD28. Numbers in outlined areas indicate percent IFN-γ+CD4+ cells. (h) IL-4 production by the cells in g. TH1, wild-type TH1 cells (negative control). Data are representative of at least three independent experiments (a–f) or two experiments (g,h; error bars (d,h), s.e.m.).
It is known that T cells that lack mTOR cannot differentiate into effector cells, in part because they have diminished responses to helper T cell–skewing cytokines9. We exposed freshly isolated T cells from wild-type, T-Rheb−/− and T Rictor−/− mice to skewing cytokines and assessed STAT activation. Consistent with their inability to become TH1 or TH17 cells, T-Rheb−/− T cells showed less phosphorylation of STAT4 and STAT3 in response to IL-12 and IL-6, respectively (Fig. 6c, left). However, T-Rheb−/− T cells demonstrated enhanced STAT6 phosphorylation in response to IL-4 (a TH2-promoting cytokine). T-Rictor−/− T cells showed intact IL-12- and IL-6-induced activation of STAT4 and STAT3, whereas IL-4-induced activation of STAT6 was diminished (Fig. 6c, right). These findings indicate that the ability of mTORC1 and mTORC2 to regulate the differentiation of effector T cells differently is due in part to their ability to regulate STAT activation differently. In response to different cytokines, Rheb-deficient T cells showed less IL-23- and IFN-β-induced activation of STAT4 and less IL-10- and IL-21-dependent activation of STAT3 (data not shown). Rictor-deficient T cells showed less STAT6 activation in response to IL-13 (data not shown).
Studies have shown that mTOR influences STAT activation9,24,25. To determine how distinct STAT proteins are regulated differently in T cells, we examined the expression of cytokine receptors on Rheb- and Rictor-deficient T cells. Wild-type, Rheb-deficient and Rictor-deficient T cells all had equivalent expression of the receptors IL-12Rβ1, IL-4R and IL-6R (Supplementary Fig. 12), which indicated that mTOR did not regulate cytokine receptor expression.
STAT activation is partially regulated by the expression of inhibitory SOCS proteins, which can dephosphorylate Jak kinase–dependent residues on STAT proteins in different ways. We assessed the expression patterns of SOCS3 and SOCS5 in the Rheb- and Rictor-deficient T cells. Consistent with published findings26,27,28, wild-type T cells expressed SOCS3 mRNA and protein at baseline and downregulated its expression after activation (Fig. 6d,e), whereas SOCS5 expression was low in naive T cells and increased with activation29,30. In resting Rheb-deficient T cells, expression of SOCS3 mRNA and protein was much higher, and these cells retained higher expression after activation (Fig. 6d,e), whereas SOCS5 expression was similar to that in wild-type cells. Rictor-deficient T cells produced SOCS3 at baseline (albeit in smaller amounts than the wild-type cells produced) and this expression decreased after activation, whereas SOCS5 expression was markedly upregulated in the resting cells and remained higher after activation (Fig. 6d,e). Thus, at baseline and after activation, SOCS3 and SOCS5 are regulated differently in Rheb-deficient and Rictor-deficient T cells.
SOCS protein expression is regulated dynamically over time. To examine the expression patterns of SOCS proteins at later time points, we activated T cells for 48, 72 or 96 h. At these time points, SOCS3 protein remained more abundant in Rheb-deficient T cells, whereas wild-type and Rictor-deficient cells downregulated SOCS3 (Fig. 6f). Furthermore, whereas wild-type and Rheb-deficient cells consistently upregulated SOCS5 after activation, Rictor-deficient T cells had much more SOCS5 at later time points.
To prove that the larger amounts of SOCS3 in the Rheb-deficient T cells and of SOCS5 in the Rictor-deficient T cells had functional importance, we transfected freshly isolated wild-type, Rheb-deficient and Rictor-deficient CD4+ T cells with small interfering RNA (siRNA) specific for SOCS3 and SOCS5. This resulted in substantial knockdown of SOCS3 and SOCS5 mRNA (Supplementary Fig. 13). In T cells stimulated in TH1 and TH2 differentiation conditions, knockdown of SOCS3 mRNA resulted in more IFN-γ production in Rheb-deficient T cells (Fig. 6g), whereas knockdown of SOCS5 mRNA in Rictor-deficient T cells resulted in a greater ability to produce IL-4 (Fig. 6h). Such observations demonstrate that SOCS proteins regulate TH1, TH17 and TH2 differentiation in the Rheb- and Rictor-deficient T cells.
Transcription factor expression in T-Rheb−/− and T-Rictor−/− T cells
Next we sought to determine if the transcription factors T-bet, RORγt and GATA-3, which have critical roles in the development of TH1, TH17 and TH2 cells, were selectively affected by deletion of Rheb or Rictor. Rheb-deficient T cells failed to fully upregulate T-bet mRNA and protein under TH1 conditions and RORγt under TH17 conditions while expressing abundant GATA-3 under TH2 conditions (Fig. 7a,b). In fact, Rheb-deficient cells had higher expression of GATA-3 than did wild-type cells, consistent with the enhanced ability of Rheb-deficient cells to phosphorylate STAT6 and produce IL-4 in vivo. In T-Rictor−/− T cells, T-bet and RORγt were transcribed and translated under TH1 and TH17 conditions, respectively, but GATA-3 was not expressed under TH2 conditions (Fig. 7a,b). In T cells stimulated in TH1 and TH2 differentiation conditions, knockdown of SOCS3 mRNA resulted in higher T-bet expression in Rheb-deficient T cells (Fig. 7c), whereas knockdown of SOCS5 mRNA in Rictor-deficient T cells resulted in higher GATA-3 expression (Fig. 7c). Furthermore, wild-type T cells exposed to a very low dose of rapamycin (which inhibits TH1 development but not TH2 development) had lower T-bet expression but not lower GATA3 expression in response to the appropriate differentiating conditions (Fig. 7d). Thus, in the absence of the nutrient-sensitive signaling pathways mediated by mTOR, the ability to respond to cytokines is diminished and T cells fail to properly upregulate lineage-specific transcription factors.

(a) Expression of mRNA for lineage-specific transcription factors in wild-type, T-Rheb−/− and T-Rictor−/− CD4+ T cells skewed toward the TH1, TH2 or TH17 subset; results are presented relative to those for 18S rRNA (T-bet and GATA-3) or as arbitrary units for the change in cycling threshold (RORγt). ND, not detected. (b) Flow cytometry of wild-type, T-Rheb−/− and T-Rictor−/− CD4+ T cells skewed toward the TH1, TH2 or TH17 subset and stained for T-bet, GATA-3 or RORγt. Numbers above outlined areas indicate percent cells in each. (c) Flow cytometry of cells transfected with siRNA (as in Fig. 6g) and skewed toward the TH1 or TH2 subset. Shaded histograms indicate wild-type TH2 cells (T-bet) or wild-type TH1 cells (GATA-3). (d) Flow cytometry of naive 5C.C7 T cells skewed toward the TH1 or TH2 subset in the presence or absence of a very low dose of rapamycin. Numbers in quadrants indicate percent GATA-3+T-bet− cells (top left) or GATA-3− T-bet+ cells (bottom right). Data are representative of at least three independent experiments (a,b,d; error bars (a), s.e.m.) or two experiments (c).
Treg cell development in the absence of mTORC1 and mTORC2
It is known that mTOR-deficient T cells differentiate down a Foxp3+ Treg cell pathway and that inhibition of mTORC1 alone is not sufficient for the generation of Treg cells9. Rapamycin can enhance the generation of inducible Treg cells31,32,33. To determine the specific effect of mTORC1 and mTORC2 on Treg cell differentiation, we cultured wild-type, T-Rheb−/−, T-Rictor−/− and T-Mtor−/− T cells in vitro under activating conditions (in the absence of exogenous transforming growth factor-β (TGF-β)) and then assessed the generation of Foxp3+ T cells. The elimination of neither mTORC1 signaling nor mTORC2 signaling alone led to more Foxp3+ cells (Fig. 8a). Notably, however, inducible Treg cells (made with TGF-β and IL-2) from wild-type, T-Rheb−/−, T-Rictor−/− mice all seemed to inhibit T cell function equally (Fig. 8b).

(a) Expression of Foxp3 and CD25 in wild-type, T-Rheb−/−, T-Rictor−/− and T-Mtor−/− CD4+ T cells stimulated overnight with anti-CD3 and APCs and allowed to 'rest' for 5 d in IL-7-supplemented medium. (b) CFSE dilution in wild-type T cells given no stimulation (No stim) or stimulated in vitro with anti-CD3 APCs alone (No supp) or in combination with Foxp3+ Treg cells (suppressor/responder (Supp/resp) ratio, above plots) generated from wild-type, T-Rheb−/− and T-Rictor−/− CD4+ T cells (with TGF-β and IL-2); Treg cells were enriched by CD25 magnetic separation, and Foxp3 expression was confirmed by intracellular staining. (c) Expression of Foxp3 and CD25 in wild-type, T-Rheb−/− and T-Rictor−/− CD4+ T cells stimulated with anti-CD3 and APCs in the presence of a very low dose or conventional (500 nM) dose of rapamycin and allowed to 'rest' for 5 d in IL-7-supplemented medium (with drug). (d) Expression of Foxp3 and CD25 (left) by wild-type T cells stimulated with anti-CD3 and APCs in the presence (DMK1) or absence (Untreated) of DMK1, followed by population expansion for 5 d in the presence of IL-2 and drug, and immunoblot analysis (right) of mTOR activation in lysates of wild-type T cells stimulated for 1 or 3 h with anti-CD3 and anti-CD28 in serum-free medium with or without DMK1. Numbers adjacent to (a,d) or in (c) outlined areas indicate percent CD25+Foxp3+ cells. Data are representative of at least three independent experiments.
To further test the hypothesis that both mTORC1 and mTORC2 must be inhibited to generate Treg cells, we activated wild-type, T-Rheb−/− and T-Rictor−/− T cells in the presence of a conventional dose of rapamycin (500 nM, which inhibits mTORC1 and mTORC2) or a very low dose of rapamycin (500 pM, which inhibits only mTORC1). Wild-type T cells differentiated into Foxp3+ T cells at the conventional dose of 500 nM (Fig. 8c). T-Rheb−/− T cells required the mTORC2 inhibition induced by the conventional dose. In contrast, T-Rictor−/− T cells require only the very low dose of rapamycin to differentiate into Treg cells. Finally, the mTOR-specific kinase inhibitor DMK1 (which acutely inhibits both mTOR downstream signaling pathways)34 promoted the generation of Treg cells from naive wild-type T cells, consistent with our data obtained with mTOR-, mTORC1- and mTORC2-deficient T cells (Fig. 8d). Thus, inhibition of both mTORC1 and mTORC2 is required for the generation of Treg cells in the absence of exogenous TGF-β.
Discussion
In this report we have defined precise and reciprocal roles for signaling by mTORC1 and mTORC2 in regulating the differentiation of CD4+ helper T cells. Our data have ascribed tissue-distinct physiological functions to mTORC1 and mTORC2. Thus, mTORC1 signaling promotes TH1 and TH17 differentiation, mTORC2 signaling promotes TH2 differentiation, and inhibition of mTOR leads to Treg cells. Preliminary studies in our laboratory have failed to demonstrate differences in the activation of mTORC1 and mTORC2 under TH1- and TH2-skewing conditions. Therefore, we hypothesize that it is the differences in expression and activation of downstream substrates and signaling pathways that confer on mTORC1 the ability to regulate TH1 and TH17 differentiation and confer on mTORC2 the ability to regulate TH2 differentiation.
Our studies provide a critical link between metabolism and T cell differentiation. It is known that mTORC1 signaling integrates growth factors, energy, oxygen and amino acids to promote processes necessary for cell growth2,5. In addition, in T cells, the PI(3)K-Akt-mTORC1 axis is activated by engagement of the coreceptor CD28 and signaling via the IL-2 receptor35. IL-7 promotes mTOR activity, helping to prevent atrophy in T cells36, whereas IL-4-induced mTOR activity prevents apoptosis37. IL-12 and IFN-γ promote the sustained activation of mTORC1 activity in CD8+ T cells11,38. Relatively little is known about the precise role of mTORC2 signaling in regulating cellular function5. The ability of mTORC2 to regulate TH2 differentiation provides potential clues for determining the specific role of mTORC2 in other cell types.
The small GTPase Rheb has been identified as an activator of mTORC1 (ref. 15). Our data have demonstrated that Rheb signaling has an important role in regulating T cell differentiation. The same block in differentiation is seen in T cells treated with a very low dose of rapamycin, T cells deficient in mTOR or T cells deficient in Rheb, which supports the hypothesis that the ability of Rheb to direct differentiation is achieved through mTORC1 activation. Furthermore, mTORC1 signaling seems to be both necessary and sufficient for TH1 and TH17 differentiation, because Rictor-deficient T cells still maintain their ability to become TH1 and TH17 effector cells.
Rictor-deficient T cells generated by crossing of mice expressing Cre from the distal promoter of the gene encoding the kinase Lck (dLck-iCre) and mice with loxP-flanked Rictor alleles have been studied39. Consistent with our study, that Rictor deletion inhibited TH2 differentiation39. However, the generation of fewer TH1 cells (albeit modest compared with TH2 inhibition) was also reported39. The cause of these divergent observations remains unclear. The differences between the two models in their Cre expression might result in subtle differences in T cell development and could affect helper T cell differentiation. Additionally, we found that Rictor-deficient T cells had a greater propensity to differentiate to TH17 cells than did wild-type cells. In addition, there seem to be differences in signaling in the Rictor-deficient T cells used in each study. Notably, unlike T cells from our mice, which demonstrated robust phosphorylation of Akt Thr308 and subsequent S6K1 activation, T cells in the other study showed less signaling upstream of mTOR39, which could also impair mTORC1 signaling. We suggest that when the signaling defects are restricted downstream of mTORC2 signaling, there is a selective block in TH2 development but not in TH1 development. Thus, it is possible that the Rictor-deficient T cells in the previously published report39 act more like mTOR-deficient T cells.
Rapamycin promotes the generation of Treg cells in the absence of exogenous TGF-β40,41,42. The ligand for the T cell–inhibitory receptor PD-1 has been shown to promote inducible Treg cells by inhibiting mTOR activation43. In addition, local depletion of essential amino acids can promote the generation of Treg cells by inhibiting mTOR activity44. We have shown here that signaling from both mTORC1 and mTORC2 must be inhibited for skewing of cells down the Treg cell pathway. Our genetic studies were complemented by a pharmacological approach. The simultaneous inhibition of mTORC1 and mTORC2 by an mTOR kinase inhibitor was a potent inducer of Treg cells in the absence of exogenous TGF-β, which indicates that drugs in this class can act as potent agents inducing immunosuppression and tolerance.
Both mTORC1 and mTORC2 can promote cell differentiation in other tissues. Cerebral cortical development is dependent on mTORC1, whereas terminal differentiation of myoblasts is mediated by mTORC2 (refs. 45,46). In this model, mTORC2 signaling inactivates the Rho-associated kinase ROCK1, which in turn promotes activation of the transcription factors Foxo1 and Foxo3a. We have demonstrated a similar role for signaling by mTORC1 and mTORC2 in regulating distinct tissue-specific transcription factors. It has been shown that rapamycin promotes the differentiation of CD8+ memory cells10. Thus, by inhibiting T-bet expression, rapamycin not only inhibits effector functions (such as IFN-γ production) but also promotes the expression of eomesodermin, which in turn promotes the generation of memory T cells11. The decrease in helper T cell–specific transcription factors is due to a decrease in specific STAT activation in response to cytokines. Although the IL-12-, IL-6- and IL-4-induced phosphorylation of STAT4, STAT3 and STAT6 was not completely lost in the mTORC1- and mTORC2-deficient T cells, the diminished response contributed to the observed differentiation failure. The ability of mTOR to regulate STAT3 phosphorylation has been described in cancer and neuron activation; however, the precise mechanism for this regulation is unclear24,25,47. We have demonstrated that in T cells, differences in the expression of SOCS3 and SOCS5 contribute to the STAT-activation defects. Along these lines, mTORC2 inhibits the activity of Foxo transcription factors7,48. Foxo proteins control expression of the transcription factor KLF2, which in turn promotes SOCS5 expression49. Preliminary studies have shown that Rictor-deficient T cells have more KLF2, consistent with the observed increase in SOCS5 in these cells (data not shown). We propose a model in which mTORC1 and mTORC2 activate distinct signaling programs, the net sum of which is to promote the differentiation of specific effector cells. Future studies should identify the mTORC1- and mTORC2-specific targets that control differentiation.
Methods
Mice.
Mice were kept in accordance with guidelines of the Johns Hopkins University Institutional Animal Care and Use Committee. C57BL/6, Cd4-Cre and OT-II mice from Jackson Laboratories were, where applicable, bred to Thy-1.1 backgrounds; 5C.C7 mice were from Taconic Farms. Mice with loxP-flanked Rheb alleles were generated in the laboratory of P.F. Worley. Mice with loxP-flanked Rictor alleles were a gift from M. Magnuson. Mice with loxP-flanked Mtor alleles were a gift from S.C. Kozma.
Antibodies and reagents.
The following antibodies for flow cytometry were from BD Biosciences: anti-B220 (RA3-6B2), anti-CD3 (2C11), anti-CD4 (RM4-5), anti-CD8 (Ly-3), anti-CD25 (PC61), anti-GATA-3 (L50-823), anti-IFN-γ (XMG1.2), anti-IL-2 (JES6-5H4), anti-IL-4 (11B11), anti-IL-5 (TRFK5), anti-IL-17A (TC11-18H10) and antibody to tumor necrosis factor (MP6-XT22). Anti-IL-5 (TRFK5), anti-RORγt (AFKJS-9), anti-T-bet (eBio4B10) and anti-Foxp3 (FJK-16s) were from eBioscience. Stimulatory anti-CD3 (2C11) and anti-CD28 (37.51), as well as neutralizing anti-IFN-γ (XMG1.2) and anti-IL-4 (11B11) were purified from hybridoma supernatants prepared 'in-house'. Neutralizing anti-IL-12p40 (C17.8) was from eBioscience. Antibody to S6K1 phosphorylated at Thr421 and Ser424 (9204), anti-S6K1(202), antibody to Akt phosphorylated at Ser473 (9271) or Thr308 (244F9), anti-Rheb1 (4935), anti-Rictor (2140), antibody to phosphorylated STAT6 (9361), anti-STAT6 (9362), antibody to phosphorylated STAT3 (D3A7) and anti-STAT3 (9132) were from Cell Signaling Technologies. Anti-Akt (H-136), antibody to phosphorylated STAT4 (sc-101804), anti-STAT4 (C-20), anti-SOCS3 (H-103) and anti-SOCS5 (M-300) were from Santa Cruz Biotechnologies. Vaccinia virus expressing ovalbumin was a gift from C. Drake. Cytokines were from Peprotech. Rapamycin was from LC Labs. DMK1 (compound 401 in ref. 34) was synthesized at Johns Hopkins. ON-TARGET siRNA reagents for control and SOCS3- or SOCS5-specific RNA-mediated interference were from Dharmacon. Nucleofection reagents were from Lonza.
Immunoblot analysis.
Immunoblot analysis was done essentially as described50. Magnetically purified CD4+ T cells were stimulated in serum-free medium with anti-CD3 (1 μg/ml), anti-CD28 (2 μg/ml) and antibody to hamster immunoglobulin G1 (0.75 μg/ml; G94-56; BD Biosciences), then were collected and lysed. For immunoblot analysis of STAT proteins, cells were kept in cytokine-free medium for 1 h, then were stimulated for 30 min with IL-12 (10 ng/ml), IL-4 (1 ng/ml) or IL-6 (10 ng/ml). For immunoblot analysis of SOCS proteins, cells were allowed to 'rest' for 2 h and then were lysed or stimulated with plate-bound anti-CD3 (5 μg/ml) and soluble anti-CD28 (2 μg/ml).
Enzyme-linked immunosorbent assay.
IL-2, IFN-γ and IL-4 were analyzed by enzyme-linked immunosorbent assay as described by the manufacturer (eBioscience).
OVA-specific antibodies were analyzed by enzyme-linked immunosorbent assay in Nunc-Immuno plates (eBioscience) coated overnight at 4 °C with 100 μl OVA (100 μg/ml in PBS) and blocked for 1 h at 25 °C with 5% (wt/vol) BSA (Roche). Wells were washed several times and serum diluted in 5% (wt/vol) BSA was added for 2 h. Wells were washed and then were incubated for 1 h with biotinylated anti-mouse immunoglobulin G1 (A85-1; BD Biosciences) diluted to a concentration of 0.5 μg/ml in 5% (wt/vol) BSA. Wells were washed and then were incubated for 30 min with streptavidin-conjugated horseradish peroxidase. Wells were washed and then were incubated for 5 min with tetramethylbenzidine substrate or until the reaction approached saturation. Linear regression analysis was used to determine titer.
T cell stimulation and skewing.
Unless otherwise stated, T cells were stimulated with anti-CD3 (5 μg/ml) and, where appropriate, anti-CD28 (2 μg/ml). APC-free systems used anti-CD3 diluted in PBS for coating of flat-bottomed plates. PMA (phorbol 12-myristate 13-acetate) and ionomycin (Sigma) were used at a concentration of 50 ng/ml and 500 ng/ml, respectively.
T cells were skewed with irradiated APCs and anti-CD3 and then washed, followed by population expansion for 5 d, then were rechallenged with plate-bound anti-CD3 and soluble anti-CD28 or with fresh APCs and anti-CD3. Sorted naive cells were skewed with plate-bound anti-CD3 and anti-CD28 and rechallenged likewise. T cells skewed after nucleofection were stimulated for 48 h, followed by population expansion for 48 h for less cell loss. Skewing conditions were as follows: TH1, IL-12 (5 ng/ml), IFN-γ (100 ng/ml) and anti-IL-4 (100 μg/ml), with IL-2 (1 ng/ml) during the rest period; TH2, IL-4 (1 ng/ml), anti-IL-12 (100 μg/ml) and anti-IFN-γ (100 μg/ml), with IL-2 (1 ng/ml) during the rest period; TH17, TGF-β (10 ng/ml), IL-6 (10 ng/ml), anti-IFN-γ (100 μg/ml) and anti-IL-4 (100 μg/ml); Treg, TGF-β (5 ng/ml) and IL-2 (1 ng/ml).
Intracellular staining.
Brefeldin A (GolgiPlug; BD Biosciences) or monensin (GolgiStop; BD Biosciences) was used for cytokine staining. Cell surfaces were stained, then cells were fixed and made permeable with BD Cytofix/Cytoperm and then stained for cytokines. Intracellular staining of transcription factors was done without stimulation, with the eBioscience Foxp3 Fixation/Permeabilization kit. Gates were set appropriately with unstimulated controls and voltages were set on the basis of isotype-matched control antibodies.
Vaccinia infection.
C57BL/6 Thy1.2+ host mice were immunized with 2 × 106 plaque-forming units of vaccinia virus expressing ovalbumin and were given adoptive transfer of 1 × 106 to 2 × 106 CD4+Thy1.1+ OT-II T cells from wild-type or T-Rheb−/− mice. Then, 4 d after transfer, splenocytes were collected and rechallenged overnight with OVA peptide (50 μg/ml; Anaspec).
OVA sensitization.
OVA (Sigma-Aldrich) was adsorbed by gentle shaking for 30 min onto Imject Alum adjuvant (Pierce) to a final concentration of 100 μg/ml. Mice were primed with 200 μl intraperitoneally and boosted 14 d later. On day 18, serum and spleens were collected. Serum was analyzed for antibodies and spleens were stimulated 48 h with OVA (100 μg/ml).
EAE.
Mice on the C57BL/6 background were immunized subcutaneously with 150 μg MOG peptide (Genway) with incomplete Freund's adjuvant (Thermo Scientific) supplemented with Mycobacterium tuberculosis (4 mg/ml; Difco). Mice received 200 μg pertussis toxin (List Biological Laboratories) intravenously on both day 0 and day 4. Mice were assigned scores as follows by a researcher 'blinded' to sample identity: 0.5, flaccid tail; 1, partial hindlimb paralysis; 2, full hindlimb paralysis; 3, partial forelimb paralysis; 4, full forelimb paralysis; 5, moribund. Mice were killed after they achieved a score of 4 or higher or at the end of the time course. The central nervous system was collected and either was frozen or infiltrates were isolated on Percoll (GE Healthcare). Infiltrates were placed in culture medium for 24 h, then stimulated for 8 h with PMA and ionomycin.
In vitro suppression.
Treg cells were generated from wild-type, T-Rheb−/− and T-Rictor−/− mice in vitro with TGF-β and IL-2, then CD25+ cells were isolated (Miltenyi-Biotec) and analyzed for Foxp3 expression by flow cytometry. Wild-type T cells were labeled with CFSE (carboxyfluorescein diacetate succinimidyl ester; Invitrogen) and then were stimulated with APCs (at a ratio of 1:10, T cells/APCs) and anti-CD3 (1 μg/ml). According to calculations from Foxp3 staining, various numbers of Treg cells were added to the stimulations to achieve the desired ratio. Suppression was assessed 72 h later by analysis of CFSE dilution by wild-type naive T cells.
RNA-mediated interference.
T cells were isolated by CD4+ negative selection and allowed to 'rest' in medium for 2 h. Cells were transfected with pools of control siRNA or SOCS3- or SOCS5-specific siRNA (200 pM; Dharmacon) with the Amaxa T cell Nucleofection kit (Lonza). T cells were resuspended at a density of 20 × 106 cells per ml in T cell nucleofection solution, added to siRNA, transfected by nucleofection with the X-001 program and immediately transferred into 1.5 ml T cell Nucleofector medium. T cells were washed after 16 h and stimulated in normal culture medium.
Change history
06 May 2011
In the version of this article initially published, the Discussion section cited a published study of mice with conditional deletion of Rictor in T cells without specifying which Lck promoter was used to drive the expression of Cre recombinase. The distal Lck promoter (dLck) was used. The error has been corrected in the HTML and PDF versions of the article.
References
Guertin, D.A. & Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 12, 9–22 (2007).
Hay, N. & Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 18, 1926–1945 (2004).
Kim, D.H. et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175 (2002).
Sarbassov, D.D. et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 14, 1296–1302 (2004).
Laplante, M. & Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 122, 3589–3594 (2009).
Holz, M.K. & Blenis, J. Identification of S6 kinase 1 as a novel mammalian target of rapamycin (mTOR)-phosphorylating kinase. J. Biol. Chem. 280, 26089–26093 (2005).
Guertin, D.A. et al. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell 11, 859–871 (2006).
Delgoffe, G.M. & Powell, J.D. mTOR: taking cues from the immune microenvironment. Immunology 127, 459–465 (2009).
Delgoffe, G.M. et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 30, 832–844 (2009).
Araki, K. et al. mTOR regulates memory CD8 T-cell differentiation. Nature 460, 108–112 (2009).
Rao, R.R., Li, Q., Odunsi, K. & Shrikant, P.A. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity 32, 67–78 (2010).
Sinclair, L.V. et al. Phosphatidylinositol-3-OH kinase and nutrient-sensing mTOR pathways control T lymphocyte trafficking. Nat. Immunol. 9, 513–521 (2008).
Yamagata, K. et al. Rheb, a growth factor- and synaptic activity-regulated gene, encodes a novel Ras-related protein. J. Biol. Chem. 269, 16333–16339 (1994).
Yee, W.M. & Worley, P.F. Rheb interacts with Raf-1 kinase and may function to integrate growth factor- and protein kinase A-dependent signals. Mol. Cell. Biol. 17, 921–933 (1997).
Manning, B.D. & Cantley, L.C. Rheb fills a GAP between TSC and TOR. Trends Biochem. Sci. 28, 573–576 (2003).
Saucedo, L.J. et al. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat. Cell Biol. 5, 566–571 (2003).
Barnden, M.J., Allison, J., Heath, W.R. & Carbone, F.R. Defective TCR expression in transgenic mice constructed using cDNA-based α- and β-chain genes under the control of heterologous regulatory elements. Immunol. Cell Biol. 76, 34–40 (1998).
Park, H. et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 6, 1133–1141 (2005).
Wensky, A.K. et al. IFN-γ determines distinct clinical outcomes in autoimmune encephalomyelitis. J. Immunol. 174, 1416–1423 (2005).
Kumar, A. et al. Muscle-specific deletion of rictor impairs insulin-stimulated glucose transport and enhances basal glycogen synthase activity. Mol. Cell. Biol. 28, 61–70 (2008).
Paul, W.E. What determines Th2 differentiation, in vitro and in vivo? Immunol. Cell Biol. 88, 236–239 (2010).
Yamane, H., Zhu, J. & Paul, W.E. Independent roles for IL-2 and GATA-3 in stimulating naive CD4+ T cells to generate a Th2-inducing cytokine environment. J. Exp. Med. 202, 793–804 (2005).
Sarbassov, D.D. et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22, 159–168 (2006).
Wang, B. et al. Nogo-66 promotes the differentiation of neural progenitors into astroglial lineage cells through mTOR-STAT3 pathway. PLoS ONE 3, e1856 (2008).
Zhou, J. et al. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl. Acad. Sci. USA 104, 16158–16163 (2007).
Yamamoto, K., Yamaguchi, M., Miyasaka, N. & Miura, O. SOCS-3 inhibits IL-12-induced STAT4 activation by binding through its SH2 domain to the STAT4 docking site in the IL-12 receptor β2 subunit. Biochem. Biophys. Res. Commun. 310, 1188–1193 (2003).
Yu, C.R. et al. Suppressor of cytokine signaling 3 regulates proliferation and activation of T-helper cells. J. Biol. Chem. 278, 29752–29759 (2003).
Palmer, D.C. & Restifo, N.P. Suppressors of cytokine signaling (SOCS) in T cell differentiation, maturation, and function. Trends Immunol. 30, 592–602 (2009).
Ozaki, A., Seki, Y., Fukushima, A. & Kubo, M. The control of allergic conjunctivitis by suppressor of cytokine signaling (SOCS)3 and SOCS5 in a murine model. J. Immunol. 175, 5489–5497 (2005).
Seki, Y. et al. Expression of the suppressor of cytokine signaling-5 (SOCS5) negatively regulates IL-4-dependent STAT6 activation and Th2 differentiation. Proc. Natl. Acad. Sci. USA 99, 13003–13008 (2002).
Battaglia, M., Stabilini, A. & Roncarolo, M.G. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood 105, 4743–4748 (2005).
Sauer, S. et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc. Natl. Acad. Sci. USA 105, 7797–7802 (2008).
Haxhinasto, S., Mathis, D. & Benoist, C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J. Exp. Med. 205, 565–574 (2008).
Ballou, L.M., Selinger, E.S., Choi, J.Y., Drueckhammer, D.G. & Lin, R.Z. Inhibition of mammalian target of rapamycin signaling by 2-(morpholin-1-yl)pyrimido[2,1-α]isoquinolin-4-one. J. Biol. Chem. 282, 24463–24470 (2007).
Colombetti, S., Basso, V., Mueller, D.L. & Mondino, A. Prolonged TCR/CD28 engagement drives IL-2-independent T cell clonal expansion through signaling mediated by the mammalian target of rapamycin. J. Immunol. 176, 2730–2738 (2006).
Rathmell, J.C., Farkash, E.A., Gao, W. & Thompson, C.B. IL-7 enhances the survival and maintains the size of naive T cells. J. Immunol. 167, 6869–6876 (2001).
Stephenson, L.M., Park, D.S., Mora, A.L., Goenka, S. & Boothby, M. Sequence motifs in IL-4Rα mediating cell-cycle progression of primary lymphocytes. J. Immunol. 175, 5178–5185 (2005).
Lekmine, F. et al. Interferon-gamma engages the p70 S6 kinase to regulate phosphorylation of the 40S S6 ribosomal protein. Exp. Cell Res. 295, 173–182 (2004).
Lee, K. et al. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity 32, 743–753 (2010).
Kang, J., Huddleston, S.J., Fraser, J.M. & Khoruts, A. De novo induction of antigen-specific CD4+CD25+Foxp3+ regulatory T cells in vivo following systemic antigen administration accompanied by blockade of mTOR. J. Leukoc. Biol. 83, 1230–1239 (2008).
Kopf, H., de la Rosa, G.M., Howard, O.M. & Chen, X. Rapamycin inhibits differentiation of Th17 cells and promotes generation of FoxP3+ T regulatory cells. Int. Immunopharmacol. 7, 1819–1824 (2007).
Valmori, D. et al. Rapamycin-mediated enrichment of T cells with regulatory activity in stimulated CD4+ T cell cultures is not due to the selective expansion of naturally occurring regulatory T cells but to the induction of regulatory functions in conventional CD4+ T cells. J. Immunol. 177, 944–949 (2006).
Francisco, L.M. et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 206, 3015–3029 (2009).
Cobbold, S.P. et al. Infectious tolerance via the consumption of essential amino acids and mTOR signaling. Proc. Natl. Acad. Sci. USA 106, 12055–12060 (2009).
Kim, S. et al. The apical complex couples cell fate and cell survival to cerebral cortical development. Neuron 66, 69–84 (2010).
Shu, L. & Houghton, P.J. The mTORC2 complex regulates terminal differentiation of C2C12 myoblasts. Mol. Cell. Biol. 29, 4691–4700 (2009).
Johnson, M.D., O'Connell, M., Vito, F. & Bakos, R.S. Increased STAT-3 and synchronous activation of Raf-1-MEK-1-MAPK, and phosphatidylinositol 3-Kinase-Akt-mTOR pathways in atypical and anaplastic meningiomas. J. Neurooncol. 92, 129–136 (2009).
Garcia-Martinez, J.M. & Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J. 416, 375–385 (2008).
Dekker, R.J. et al. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood 107, 4354–4363 (2006).
Delgoffe, G.M., Kole, T.P., Cotter, R.J. & Powell, J.D. Enhanced interaction between Hsp90 and raptor regulates mTOR signaling upon T cell activation. Mol. Immunol. 46, 2694–2698 (2009).
Acknowledgements
We thank P.F. Worley (Johns Hopkins University) for mice with loxP-flanked Rheb alleles; M. Magnuson (Vanderbilt University) for mice with loxP-flanked Rictor alleles; S.C. Kozma (University of Cincinnati) for mice with loxP-flanked Mtor alleles; C. Drake (Johns Hopkins University) for vaccinia virus expressing ovalbumin; members of the Powell laboratory; and C. Drake and D. Pardoll for discussions and reagents. Supported by the US National Institutes of Health (R01AI077610-01A2).
Author information
Authors and Affiliations
Contributions
G.M.D. did research, helped design experiments and wrote the paper; K.N.P. assisted with in vivo experiments and biochemistry; A.T.W. assisted with EAE induction and central nervous system isolation and did immunohistochemistry; E.H. assisted with very-low-dose rapamycin experiments; D.J.M. synthesized the mTOR kinase inhibitor; M.R.H. helped design experiments and contributed reagents; B.X. and P.F.W. generated the original mouse line with loxP-flanked Rheb alleles; and J.D.P. designed experiments, oversaw research and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–13 (PDF 9152 kb)
Supplementary Video 1
Rheb-deficient T cells induce nonclassical EAE. Video of a representative T-Rheb−/− mouse 14 days after immunization with MOG peptide + CFA to induce EAE. The mouse displays symptoms of ataxia without paralysis. (MOV 5111 kb)
Rights and permissions
About this article
Cite this article
Delgoffe, G., Pollizzi, K., Waickman, A. et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol 12, 295–303 (2011). https://doi.org/10.1038/ni.2005
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ni.2005
This article is cited by
-
Mechanistic target of rapamycin (mTOR): a potential new therapeutic target for rheumatoid arthritis
Arthritis Research & Therapy (2023)
-
The SMYD3-dependent H3K4me3 status of IGF2 intensifies local Th2 differentiation in CRSwNP via positive feedback
Cell Communication and Signaling (2023)
-
Breast feeding, obesity, and asthma association: clinical and molecular views
Clinical and Molecular Allergy (2023)
-
Tumor-secreted IFI35 promotes proliferation and cytotoxic activity of CD8+ T cells through PI3K/AKT/mTOR signaling pathway in colorectal cancer
Journal of Biomedical Science (2023)
-
mTORC2 acts as a gatekeeper for mTORC1 deficiency-mediated impairments in ILC3 development
Acta Pharmacologica Sinica (2023)
