
Abstract
The oncogenic mechanisms underlying acute lymphoblastic leukemia (ALL) in adolescents and young adults (AYA; 15–39 years old) remain largely elusive1,2,3. Here we have searched for new oncogenes in AYA-ALL by performing RNA-seq analysis of Philadelphia chromosome (Ph)-negative AYA-ALL specimens (n = 73) with the use of a next-generation sequencer. Interestingly, insertion of D4Z4 repeats containing the DUX4 gene into the IGH locus was frequently identified in B cell AYA-ALL, leading to a high level of expression of DUX4 protein with an aberrant C terminus. A transplantation assay in mice demonstrated that expression of DUX4-IGH in pro-B cells was capable of generating B cell leukemia in vivo. DUX4 fusions were preferentially detected in the AYA generation. Our data thus show that DUX4 can become an oncogenic driver as a result of somatic chromosomal rearrangements and that AYA-ALL may be a clinical entity distinct from ALL at other ages.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

Accession codes
Change history
29 August 2016
In the version of this article initially published, the following statement was not included in the Online Methods: "The results published here are in part based upon data generated by the TARGET initiative managed by the National Cancer Institute (NCI). The RNA-seq data set and clinical information for the TARGET ALL project used for this analysis are available at the database of Genotypes and Phenotypes (dbGaP) under accession phs000218.v13.p3. We also used RNA-seq data from normal blood cells (dbGaP phs000424.v6.p1) generated as part of the GTEx project." The error has been corrected in the HTML and PDF versions of the article.
References
Harrison, C.J. Cytogenetics of paediatric and adolescent acute lymphoblastic leukaemia. Br. J. Haematol. 144, 147–156 (2009).
Burke, P.W. & Douer, D. Acute lymphoblastic leukemia in adolescents and young adults. Acta Haematol. 132, 264–273 (2014).
Wood, W.A. & Lee, S.J. Malignant hematologic diseases in adolescents and young adults. Blood 117, 5803–5815 (2011).
Hayakawa, F. et al. Markedly improved outcomes and acceptable toxicity in adolescents and young adults with acute lymphoblastic leukemia following treatment with a pediatric protocol: a phase II study by the Japan Adult Leukemia Study Group. Blood Cancer J. 4, e252 (2014).
McPherson, A. et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput. Biol. 7, e1001138 (2011).
Den Boer, M.L. et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 10, 125–134 (2009).
Roberts, K.G. et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell 22, 153–166 (2012).
Clapp, J. et al. Evolutionary conservation of a coding function for D4Z4, the tandem DNA repeat mutated in facioscapulohumeral muscular dystrophy. Am. J. Hum. Genet. 81, 264–279 (2007).
Cabianca, D.S. & Gabellini, D. The cell biology of disease: FSHD: copy number variations on the theme of muscular dystrophy. J. Cell Biol. 191, 1049–1060 (2010).
Snider, L. et al. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet. 6, e1001181 (2010).
Wijmenga, C. et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat. Genet. 2, 26–30 (1992).
Dixit, M. et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc. Natl. Acad. Sci. USA 104, 18157–18162 (2007).
Jones, T.I. et al. Facioscapulohumeral muscular dystrophy family studies of DUX4 expression: evidence for disease modifiers and a quantitative model of pathogenesis. Hum. Mol. Genet. 21, 4419–4430 (2012).
Kowaljow, V. et al. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul. Disord. 17, 611–623 (2007).
Geng, L.N. et al. DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev. Cell 22, 38–51 (2012).
Gocho, Y. et al. A novel recurrent EP300-ZNF384 gene fusion in B-cell precursor acute lymphoblastic leukemia. Leukemia 29, 2445–2448 (2015).
Martini, A. et al. Recurrent rearrangement of the Ewing's sarcoma gene, EWSR1, or its homologue, TAF15, with the transcription factor CIZ/NMP4 in acute leukemia. Cancer Res. 62, 5408–5412 (2002).
Yuki, Y. et al. Identification of a novel fusion gene in a pre-B acute lymphoblastic leukemia with t(1;19)(q23;p13). Cancer Sci. 95, 503–507 (2004).
Prima, V. & Hunger, S.P. Cooperative transformation by MEF2D/DAZAP1 and DAZAP1/MEF2D fusion proteins generated by the variant t(1;19) in acute lymphoblastic leukemia. Leukemia 21, 2470–2475 (2007).
Tsuzuki, S. & Seto, M. TEL (ETV6)-AML1 (RUNX1) initiates self-renewing fetal pro-B cells in association with a transcriptional program shared with embryonic stem cells in mice. Stem Cells 31, 236–247 (2013).
Russell, L.J. et al. IGH@ translocations are prevalent in teenagers and young adults with acute lymphoblastic leukemia and are associated with a poor outcome. J. Clin. Oncol. 32, 1453–1462 (2014).
Roth, D.B. Restraining the V(D)J recombinase. Nat. Rev. Immunol. 3, 656–666 (2003).
Corona, E.D., Jacquelin, D., Gatica, L. & Rosa, A.L. Multiple protein domains contribute to nuclear import and cell toxicity of DUX4, a candidate pathogenic protein for facioscapulohumeral muscular dystrophy. PLoS One 8, e75614 (2013).
Kawamura-Saito, M. et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum. Mol. Genet. 15, 2125–2137 (2006).
Schroeder, A. et al. The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 7, 3 (2006).
Corveleyn, A. et al. Cellular transformation of NIH3T3 fibroblasts by CIZ/NMP4 fusions. J. Cell. Biochem. 94, 1112–1125 (2005).
Acknowledgements
We thank M. Tamura and A. Maruyama for technical assistance. We are also grateful to H. Yoshimi and S. Yamazaki for providing leukemia specimens. This study was supported in part by grants for Leading Advanced Projects for Medical Innovation (LEAP) (to H.M.), for Programs for Development of Innovative Research on Cancer Therapeutics (P-DIRECT) (to T.N.) and for Practical Research for Innovative Cancer Control (to H.M. and F.H.) from the Japan Agency for Medical Research and Development. A list of centers and physicians that participated in the ALL202-U study and that provided clinical specimens can be found in the Supplementary Note.
Author information
Authors and Affiliations
Contributions
T.N. and H.M. conceived and headed the project. F.H., T.S., T. Yujiri, E.K., K.F., Y.U., Y.A., S.O., J.T., M. Kurokawa, M.T., H.K. and Y.M. collected clinical specimens and performed clinical analyses. T. Yasuda, M. Kawazu, A.K., K.D., S.M. and M.F. performed RNA-seq and genomic analyses. T. Yasuda, S.T., M. Kawazu, F.H., N.I., S. Kohsaka, E.S. and H.K. conducted functional assays. S. Kojima, K.D., T.U. and S.M. carried out computational analyses. H.M. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Screening of fusion genes by RNA-seq data.
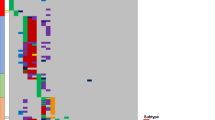
(a) Schematic of the structure of the proteins encoded by novel fusion genes detected in the screening cohort. Amino acid numbers are indicated below each structure. (b) The 54 cases of B cell AYA-ALL in the screening cohort plus 3 Ph+ ALL cases (indicated by asterisks) were clustered according to the expression values for 187 genes9 (Supplementary Table 6) with the use of Ward correlation algorithms. Patients were clustered into two large branches, one of which contains Ph+ cases as well as those with CRLF2 fusions (Ph-like group). Patient ID and fusion genes are depicted at the bottom. The expression level of each gene is color-coded. (c) Survival curves are shown for the Ph-like and non-Ph-like groups. The P value was determined with the log-rank test. (d) The 47 patients with non-Ph-like AYA-ALL were clustered with the group average method according to the expression level of the top 1% of genes (n = 237) in the rank-ordered gene list for the s.d. value of gene expression. Individuals harboring DUX4, ZNF384 or MEF2D fusions are differentially colored.
Supplementary Figure 2 Genomic rearrangements involving D4Z4 repeats.
(a) The entire translocated segment containing DUX4 was amplified by PCR from the genomic DNA of leukemic blasts from patient JALSG-002 and was then subjected to Sanger sequencing. A fragment of mitochondrial DNA (Chr. M) and two copies of D4Z4, flanked by unknown sequences, was inserted at a position between IGHJ6 and IGHV3-15. (b) A similar analysis to that in a was conducted for leukemic blasts from patient JALSG-006. A fragment of chromosome 15 and one copy of D4Z4, flanked by unknown sequences, was inserted at a position between IGHJ4 and IGHV3-66. (c) The structures of the genomic regions of D4Z4 translocation in JALSG-004, JALSG-006, TUH-005 and TUH-007 are schematically shown as in Figure 2a. (d) The fusion point between DUX4 and IGH was amplified by PCR from the genomic DNA of bone marrow mononuclear cells from patient TUH-005 before treatment (Primary), at the first complete remission (CR), the first relapse and the second complete remission. The fusion point of DUX4-MIR3697 was similarly analyzed for patient TUH-007 at the first relapse of leukemia and at the second complete remission for a duration of 3 or 7 months. A no-template control (–) was included in each analysis, and ACTB was amplified as an internal control.
Supplementary Figure 3 Functional characterization of DUX4 fusion proteins.
(a) The protein structure of the DUX4 fusions is shown as in Figure 2c. The C termini of the DUX4 fusion protein from patients JALSG-007 and TUH-005 are encoded by a genomic fragment from IGH, whereas those for patients JALSG-002, JALSG-006, JALSG-008, JALSG-009, JALSG-010 and TUH-007 are encoded by a genomic fragment other than the IGH locus (Non-IGH). Amino acid numbers are indicated below each structure. (b) The abundance of DUX4 mRNA normalized by that of ACTB mRNA was determined by TaqMan RT-PCR analysis for 16 normal human tissues (leukocytes, lung, placenta, small intestine, colon, liver, spleen, heart, ovary, kidney, skeletal muscle, prostate, pancreas, brain, thymus and testis) as well as for 5 B cell ALL lines and 90 B cell ALL specimens from the screening and validation cohorts. The abundance of DUX4 was further normalized with that of normal testis. Data are shown as means + s.d. for three independent experiments. The asterisk denotes the NALM6 cell line. (c) The total cell lysates of NIH3T3 cells expressing DUX4 or DUX4-IGH fusion proteins as in Figure 3a were immunoblotted with antibody to DUX4-N or ACTB. NIH3T3 cells infected with an empty virus (Mock) were similarly analyzed. The asterisks indicate non-specific bands with antibody to DUX4-N. (d) The same NIH3T3 cells in c were stained with Hoechst 33258, and the number of nuclei was counted in a surface area of 90,000 μm2. Data are shown as means + s.d. for three independent experiments. The P values for the indicated comparisons were determined with the Welch’s t test. (e) Mouse NIH3T3 cells were infected with a recombinant retrovirus expressing GFP alone (Mock) or GFP with DUX4 or the DUX4-IGH fusions identified in patients with ALL. The GFP+ cell fraction was evaluated by flow cytometry at the indicated times. Data were normalized by the value for day 2 and are shown as means ± s.d. for three independent experiments (left). Note that only wild-type DUX4 markedly suppressed cell growth. Right, HEK293T cells (1 × 105) were transiently transfected with 600 ng of expression plasmid for wild-type DUX4 or the DUX4 fusions, and the enzymatic activity of CASP3 and CASP7 was measured at 36 h after transfection with the Caspase-Glo 3/7 Assay. Enzymatic activities were normalized by that of the mock-transfected cells. Data are shown as means + s.d. of three independent experiments. The P value was calculated by Welch’s t test. As shown here, only wild-type DUX4 induces CASP3 and CASP7 activities.
Supplementary Figure 4 DUX4 in NALM6.
(a) The entire translocated segment containing DUX4 was amplified by PCR from the genomic DNA of NALM6 cells. Sanger sequencing of the PCR product demonstrated translocation of two copies of D4Z4 flanked by unknown sequences to a position between IGHJ4 and IGHD2-15. (b) Long-read sequencing of the NALM6 genome. Top, the structure of the D4Z4 translocation region. Middle, three SMRT subreads that fully or partly cover the rearranged region are illustrated. Bottom, three dot plots showing that the fused sequence is supported by each of the three SMRT subreads, confirming the presence of the fused sequence. (c) The 3′ end of the DUX4-IGH mRNA was determined by 3′ RACE with mRNA isolated from NALM6 cells. The sequencing electrophoretogram shows the presence of a putative polyadenylation signal (ATTAAA; red box) derived from the IGHD2-15 sequence followed by a poly(A) sequence. (d) Total cell lysates prepared from NALM6 cells infected with retroviruses encoding DUX4-IGH shRNA (DUX4-sh) or a control shRNA (Control-sh) were subjected to immunoblot analysis with antibody to DUX4-C or ACTB. (e) CCRF-SB cells were infected with recombinant retroviruses encoding both GFP and either a control shRNA (Control-sh) or DUX4-IGH shRNA (DUX4-sh). The GFP+ cell fraction was evaluated by flow cytometry after incubation of the cells for the indicated times. Data were normalized by the value for day 3 and are shown as means ± s.d. for three independent experiments.
Supplementary Figure 5 Structures of ZNF384 and MEF2D fusion proteins.
(a) The protein structures of various ZNF384 fusions are shown schematically, with amino acid numbers indicated below. (b) An RT-PCR product encompassing the fusion point of SYNRG-ZNF384 mRNA from patient JALSG-019 was subjected to Sanger sequencing, demonstrating an in-frame fusion between exon 14 of SYNRG and exon 3 of ZNF384. (c) The protein structures of various MEF2D fusions are shown schematically, with amino acid numbers indicated below. (d) Genomic DNA from the bone marrow of patient NUH-004 before chemotherapy (Primary) or at complete remission (CR) was subjected to PCR to amplify the fusion point of EP300-ZNF384 (upper left). RT-PCR was conducted for patient NUH-026 at various clinical stages to amplify the fusion point of EP300-ZNF384 (upper right). The fusion point of MEF2D-BCL9 was similarly amplified from genomic DNA and RNA from patients TUH-004 and TUP-014, respectively. A no-template control (–) was included in each analysis, and ACTB was amplified as an internal control. Primer sequences are shown in Supplementary Table 11.
Supplementary Figure 6 Transforming ability of ZNF384 and MEF2D fusions.
(a) Total cell lysates prepared from HEK293T cells transfected with expression vectors for wild-type ZNF384, EP300-ZNF384 or SYNRG-ZNF384 were subjected to immunoblot analysis with antibodies to ZNF384 and ACTB. Total cell lysates of the mock-transfected HEK293T cells were similarly analyzed. (b) A reporter plasmid for ZNF384, in which expression of firefly luciferase is driven by the human MMP7 promoter, was transiently introduced into HEK293T cells together with the internal control plasmid pGL-TK and expression plasmids for wild-type ZNF384, EP300-ZNF384 and SYNRG-ZNF384 (or the corresponding empty vector; Mock). Luciferase activities were measured 2 d after transfection, and the activity of firefly luciferase was normalized by that of Renilla reniformis luciferase. Data are shown as means + s.d. for three independent experiments. (c) Total cell lysates prepared from HEK293T cells transfected with expression vectors for wild-type MEF2D, MEF2D-BCL9 or MEF2D-HNRNPUL1 were subjected to immunoblot analysis with antibodies to MEF2D and ACTB. Total cell lysates of the mock-transfected HEK293T cells were similarly analyzed. (d) A reporter plasmid for MEF2D was transiently introduced into HEK293T cells together with the internal control plasmid pGL-TK and expression plasmids for wild-type MEF2D, MEF2D-BCL9 and MEF2D-HNRNPUL1 (or the corresponding empty vector; Mock). Luciferase activities were measured as in b. Data are shown as means + s.d. for three independent experiments. (d) Mouse NIH3T3 fibroblasts were infected with recombinant retroviruses encoding wild-type ZNF384, EP300-ZNF384, SYNRG-ZNF384, wild-type MEF2D, MEF2D-BCL9 or MEF2D-HNRNPUL1 or with the corresponding empty virus (Mock). The infected cells were cultured for 12 d in the presence of 5% bovine serum and then examined by phase-contrast microscopy. Scale bars, 100 μm.
Supplementary Figure 7 In vivo expansion of pro-B cells expressing the fusion genes.
(a) Mouse pro-B cells were infected with recombinant retroviruses encoding GFP and DUX4-IGH (n = 10), EP300-ZNF384 (n = 6), MEF2D-BCL9 (n = 15) or wild-type DUX4 (n = 7) or the corresponding empty virus (Mock; n = 9) and were then transplanted into sublethally irradiated immunocompromised mice. At 28 d after transplantation, the GFP+ fraction of bone marrow cells was quantified by flow cytometry and expressed as a ratio relative to the fraction in the original infected pro-B cells. Data are presented as box-and-whisker plots. The P values for the indicated comparisons were determined with the Mann-Whitney U test. Note that expression of wild-type DUX4 was driven by the IRES fragment. (b) The bar graphs show the percentage of the GFP+ fraction for the surface markers analyzed with flow cytometry in Figure 4a,c,d.
Supplementary Figure 8 Differentiation arrest induced by the fusion genes.
(a) Expression plasmids for FLAG epitope–tagged DUX4-IGH (identified in patient JALSG-003) and its truncation mutants (top) were introduced into HEK293T cells that were subjected to immunoblot analysis with antibody to DUX4-C or FLAG. Note that antibody to DUX4-C can recognize DUX4-IGH from patient JALSG-003 and that the epitope for the antibody presumably maps to amino acids 371–399 of DUX4. (b) The GFP+ pro-B cell fractions of bone marrow from mice transplanted with DUX4-IGH–expressing pro-B cells were subjected to immunoblot analyses with antibody to DUX4-C or tubulin (left). Total cell lysates of NALM6 cells and pro-B cells from leukemic mice were similarly examined (right). (c) The GFP+ fraction of bone marrow from mice transplanted with infected pro-B cells as in Supplementary Figure 7 was evaluated by flow cytometry at 28 d after transplantation. For recipients with ≥1% GFP+B220+ cells in bone marrow, the cell surface expression of the indicated differentiation markers was measured by flow cytometry. The P values for the indicated comparisons were determined with the Mann-Whitney U test. (d) Mouse pro-B cells infected with a recombinant retrovirus encoding GFP alone (Mock) or GFP plus DUX4 were transplanted into irradiated immunocompromised mice. At 28 d after transplantation, the B220+GFP+ fraction of bone marrow was examined for the cell surface expression of differentiation markers by flow cytometry.
Supplementary Figure 9 In vivo effects of the fusion genes.
(a) Bone marrow (BM) cells, spleen and liver were isolated after mice transplanted with pro-B cells expressing DUX4-IGH (left) or EP300-ZNF384 (right) had developed leukemia. Genomic DNA prepared from the samples was subjected to PCR to amplify the immunoglobulin D-J gene region as previously described (Immunity 12, 441–450, 2000). PCR was also performed with pro-B cells before retrovirus infection. Note that expansion of B cells with monoclonal D-J recombination was apparent in all tested organs of the leukemic mice. (b) Survival probability of the secondarily transplanted mice with DUX4-IGH+ pro-B cells (left). Note that all mice (n = 4) died within 22 d of transplantation. May-Grünwald Giemsa staining of the CytoSpin preparations of bone marrow as well as spleen from those mice demonstrated that such organs were filled with immature blasts (right).
Supplementary Figure 10 Copy number analysis of the fusion genes.
The relative gene dosage of DUX4-IGH, EP300-ZNF384 or MEF2D-BCL9 with respect to the corresponding wild-type gene was examined with droplet digital PCR (QX200, Bio-Rad) for the patients indicated below. Data are shown as means ± s.d. of three independent experiments, and the primer sequences are shown in Supplementary Table 12. Note that karyotype analyses indicated that chromosome number was 46 or 47 for the leukemic blasts of all patients (Supplementary Tables 1 and 4). The fact that the gene dosage of the fusion genes is mostly 0.8–1.0 indicates, therefore, that the allele frequency of the fusion genes is 40–50%.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–10 and Supplementary Note. (PDF 4989 kb)
Supplementary Tables 1–13
Supplementary Tables 1–13 (XLSX 84 kb)
Rights and permissions
About this article

Cite this article
Yasuda, T., Tsuzuki, S., Kawazu, M. et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat Genet 48, 569–574 (2016). https://doi.org/10.1038/ng.3535
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3535
This article is cited by
-
Promoter swapping of truncated PDGFRB drives Ph-like acute lymphoblastic leukemia
npj Precision Oncology (2023)
-
Gastrointestinal cancer occurs as extramuscular manifestation in FSHD1 patients
Journal of Human Genetics (2023)
-
EBF1–JAK2 inhibits the PAX5 function through physical interaction with PAX5 and kinase activity
International Journal of Hematology (2023)
-
International Consensus Classification of acute lymphoblastic leukemia/lymphoma
Virchows Archiv (2023)
-
Clinical characteristics and outcomes of B-cell precursor ALL with MEF2D rearrangements: a retrospective study by the Ponte di Legno Childhood ALL Working Group
Leukemia (2023)
