
Abstract
The Brassica genus encompasses three diploid and three allopolyploid genomes, but a clear understanding of the evolution of agriculturally important traits via polyploidy is lacking. We assembled an allopolyploid Brassica juncea genome by shotgun and single-molecule reads integrated to genomic and genetic maps. We discovered that the A subgenomes of B. juncea and Brassica napus each had independent origins. Results suggested that A subgenomes of B. juncea were of monophyletic origin and evolved into vegetable-use and oil-use subvarieties. Homoeolog expression dominance occurs between subgenomes of allopolyploid B. juncea, in which differentially expressed genes display more selection potential than neutral genes. Homoeolog expression dominance in B. juncea has facilitated selection of glucosinolate and lipid metabolism genes in subvarieties used as vegetables and for oil production. These homoeolog expression dominance relationships among Brassicaceae genomes have contributed to selection response, predicting the directional effects of selection in a polyploid crop genome.
Similar content being viewed by others

Main
The Brassica genus contains a diverse range of oilseed and vegetable crops important for human nutrition1. Crops of particular agricultural importance include three diploid species, Brassica rapa (AA), Brassica nigra (BB) and Brassica oleracea (CC), and three allopolyploid species, B. napus (AACC), B. juncea (AABB) and Brassica carinata (BBCC). The evolutionary relationships among these Brassica species are described by what is called the 'triangle of U' model2, which proposes how the genomes of the three ancestral Brassica species, B. rapa, B. nigra and Brassica oleracae, combined to give rise to the allopolyploid species of this genus. B. juncea formed by hybridization between the diploid ancestors of B. rapa and B. nigra, followed by spontaneous chromosome doubling. Subsequent diversifying selection then gave rise to the vegetable- and oil-use subvarieties of B. juncea. These subvarieties include vegetable and oilseed mustard in China, oilseed crops in India, canola crops in Canada and Australia, and condiment crops in Europe and other regions3. Cultivation of B. juncea began in China about 6,000 to 7,000 years ago4, and flourished in India from 2,300 BC onward5.
The genomes of B. rapa, B. oleracea and their allopolyploid offspring B. napus have been published recently6,7,8, and are often used to explain genome evolution in angiosperms6,7,8. The genomes of all Brassica species underwent a lineage-specific whole-genome triplication6,7,9, followed by diploidization that involved substantial genome reshuffling and gene losses6,10,11,12,13. In general, plant genomes are typically repetitive, polyploid and heterozygous, which complicates genome assembly14. The short read lengths of next-generation sequencing hinder assembly through complex regions, and fragmented draft and reference genomes usually lack skewed (G+C)-content sequences and repetitive intergenic sequences. Furthermore, in allopolyploid species, homoeolog expression dominance or bias, and specifically differential homoelog gene expression, has often been detected, for instance in Gossypium15,16,17, Triticum18,19 and Arabidopsis20,21, but the role of this phenomenon in selection for phenotypic traits remains mechanistically mysterious22.
We reported here the draft genomes of an allopolyploid, B. juncea var. tumida, constructed by de novo assembly using shotgun reads, single-molecule long reads (PacBio sequencing), genomic (optical) mapping (BioNano sequencing) and genetic mapping, serving to resolve complicated allopolyploid genomes. The multiuse allopolyploid B. juncea genome offers a distinctive model to study the underlying genomic basis for selection in breeding improvement. These findings place this work into the broader context of plant breeding, highlighting a potential link between homoeolog expression dominance and trait improvement that might be extendable to other polyploid crops.
Results
Genome assembly, scaffold anchoring and annotation
To distinguish among subgenomes in Brassica species, we redesignated the subgenomes in Brassica23 as follows: B. rapa as BraA; B. nigra as BniB; B. oleracea as BolC; B. juncea A subgenome as BjuA and B subgenome as BjuB; and B. napus A subgenome as BnaA and C subgenome as BnaC.
We selected an advanced generation inbred line of B. juncea var. tumida (variety T84−66) for whole-genome sequencing. We estimated the size of the T84-66 genome at 922 Mb by flow cytometry (Supplementary Fig. 1 and Supplementary Table 1). We assembled the T84-66 genome using 176× Illumina shotgun reads and 12× PacBio single-molecule long reads (Supplementary Table 2a,b and Supplementary Fig. 2). The assembly spanned 784 Mb, 85% of the 922 Mb estimated by flow cytometry (Supplementary Table 3). The contig N50 value was 61 kb, and the scaffold N50 was 855 kb (Supplementary Table 3).
We collected 996,648 BioNano DNA molecules over 150 kb, which corresponds to 222 equivalents of the genome, the average of which exceeded 2 Mb in size (Supplementary Table 4). The genome map assembled de novo consisted of 922 constituent genome maps with average length of 1.19 Mb and N50 of 1.84 M (Supplementary Table 4). We used these assemblies to correct the genome assembly above (Supplementary Fig. 3). The final assembly by the BioNano approach spanned 955 Mb, and the scaffold N50 was 1.5 Mb (Supplementary Table 3). We constructed a high-resolution genetic map with 5,333 bin markers and 18 pseudo-chromosomes (10A and 8B subgenomes; Supplementary Tables 5 and 6). We then integrated a published B. juncea genetic map24 (Supplementary Table 7). Finally, we anchored 91.5% and 72.3% of A- and B-subgenome assembly sequences onto the 10 and the 8 pseudo-chromosomes, respectively (Supplementary Table 8a and Supplementary Fig. 4). We sorted the B. juncea chromosomes into the 402.1 Mb BjuA and 547.5 Mb BjuB subgenomes based on this assembly (Supplementary Table 9).
We also sequenced the genome of a doubled haploid line of B. nigra (YZ12151) for comparative genomic study. We assembled a collection of 96× Illumina shotgun reads to generate a 396.9 Mb genome sequence for B. nigra, with a scaffold N50 of 557.3 kb, and 68% of the estimated 591 Mb B. nigra genome (Supplementary Tables 10 and 11, and Supplementary Fig. 5). We anchored the 66% scaffolds into pseudo-chromosomes for B. nigra, referring to the BjuB genetic map (Supplementary Table 8b).
To validate the genome assembly, we used subreads from PacBio, of which 10 subreads had more than 99.4% coverage and 92.3% identity, on average, with the assembled genome (Supplementary Table 12). We aligned 15 published bacterial artificial chromosomes (BACs) from B. nigra to the B. nigra genome assembly, and observed over 98.5% coverage and 99.8% identity on average to BAC clones (Supplementary Table 13 and Supplementary Fig. 6). We BLAST-aligned 458 core eukaryotic genes (Cluster of Essential Genes (CEG) database)25 to the genome assembly with core eukaryotic genes mapping approach (CEGMA) pipeline26, which showed high-confidence hits of 453 (98.8%) and 458 (100%) CEG proteins for all 458 essential genes in CEG with full length (>70% alignment) in the genome of B. juncea and B. nigra, respectively (Supplementary Table 14a). We validated the assembled genomes by matching expressed sequence tags (ESTs) downloaded from the US National Center for Biotechnology Information (NCBI) database, which indicated that 98.9% and 98.2% ESTs were supported by the assembled genomes of B. juncea and B. nigra (>50% alignments), respectively (Supplementary Table 14b).
We identified and compared repetitive sequences from syntenic regions of these genomes. We identified 316.1 Mb of repetitive sequence from the B. juncea genome, 131.2 Mb from BjuA and 216.5 Mb from BjuB (Supplementary Table 15). Long terminal repeats (LTRs) are the predominant transposable element (TE) family identified in all sequenced Brassica genomes6,7. Copia- and Gypsy-type LTRs represent the two most abundant TE subfamilies. Using repetitive sequence from syntenic regions, we found that they constituted a similar percentage of all TEs in the BjuA and BjuB, and their respective ancestral genomes (Supplementary Fig. 7a). We observed similar repetitive sequence contributions in B. napus (Supplementary Fig. 7b). We identified TEs in the B. juncea and B. napus subgenomes that were newly formed after divergence from each ancestral genome (Supplementary Fig. 8 and Supplementary Table 16a). We confirmed five randomly selected newly formed TEs by PCR amplification from B. rapa, B. nigra and B. juncea (Supplementary Fig. 9). These newly formed TEs showed similar distribution and percentage between the B. juncea and B. napus subgenomes, and their respective ancestral genomes (Supplementary Table 16b and Supplementary Fig. 10a,b). We observed 310 newly formed TEs to be active between the subgenomes of B. juncea, a much larger number than the 41 newly formed TEs being found active between the subgenomes of B. napus (Supplementary Table 17).
We annotated 80,050 and 49,826 protein-coding genes in the B. juncea and B. nigra genomes, respectively (Supplementary Table 18). Approximately 97.8% of B. juncea genes and 94.7% of B. nigra genes could be annotated by non-redundant nucleotide and protein sequences in the NCBI, Cluster of Orthologous Groups (COG), Gene Ontology (GO), SWISS-PROT and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases (Supplementary Table 19). Using transcriptomes of B. juncea we validated gene predictions of assembled genomes, verifying all predictions (Supplementary Table 20a,b). Additionally, we identified noncoding RNAs, consisting of 21 long noncoding RNAs, 3,725 small RNAs, 2,638 tRNAs, 511 rRNAs, 1,402 microRNAs and 15,418 small nuclear RNAs, from the B. juncea genome (Supplementary Table 21).
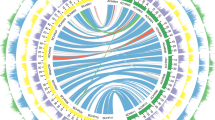
We extracted 28,228 and 28,917 syntenic ortholog gene pairs from the B. juncea subgenomes and their ancestral genomes to identify gene loss during the speciation process8. In total, we identified 562 and 545 genes lost from BjuA and BjuB, respectively, relative to their common ancestral genomes. This represents a higher percentage than the gene loss estimates for BnaA and BnaC, relative to their common ancestral genomes (Supplementary Table 22). We validated gene loss using PCR amplifications (Supplementary Fig. 11). Gene loss numbers of B. juncea and B. napus were consistent with their formation times. The identified genes lost in the B. juncea subgenomes of BjuA and BjuB are involved in different functions based on Gene Ontology (Supplementary Fig. 12a,b). We mapped the distributions of genes, repetitive sequences, gene loss, pseudogenes, genome markers and genetic markers of the B. juncea subgenomes (Fig. 1).

The B. juncea genome comprises 10 chromosomes belonging to BjuA (J01−J10; right semicircle) and 8 chromosomes belonging to BjuB (J11−J18; left semicircle), scaled on the basis of their assembled length. Homeologous relationships between BjuA and BjuB chromosomes are displayed with connecting lines colored according to the BjuB subgenome. The tracks, from outer to inner, show gene density (non-overlapping, window size = 500 kb), repetitive sequence density (window size = 500 kb), the location of gene loss (blue solid point for gene loss), the location of pseudogenes (solid line for pseudogenes), genome (optical) marker density (window size = 500 kb) and genetic marker density (window size = 500 kb).
Comparison of A subgenomes in Brassica
Synteny analysis among three A subgenomes of Brassica showed strong co-linearity, although chromosomal rearrangements have occurred between BjuA and BraA after their divergence from the common B. rapa ancestor (Fig. 2a and Supplementary Table 23). To study the divergence of BjuA and BnaA, we assayed single-nucleotide polymorphisms (SNPs) from the resequencing of A subgenomes from eighteen B. juncea accessions including the B. juncea reference sequence (Supplementary Table 24), five B. napus accessions including the B. napus reference sequence8, and 27 B. rapa accessions, including the B. rapa reference sequence6 that covers most subspecies of B. rapa (Supplementary Table 25). We constructed a neighbor-joining tree for A subgenomes in Brassica, and discovered that BjuA and BnaA had divergent origins (Fig. 2b). BjuA might derive from B. rapa ssp. tricolaris, which is distributed in Asia, whereas BnaA might derive from B. rapa ssp. rapa (European turnip), which is widely distributed in Europe (Fig. 2b). This discovery indicates that allopolyploids B. juncea and B. napus have independent geographical origins, deriving from Asian and European regions, respectively.

(a) Schematic representation of synteny among B. rapa (BraA), A subgenome of B. juncea (BjuA) and B. napus (BnaA). Each line connects a pair of orthologous genes between genomes or subgenomes. (b) A phylogenetic neighbor-joining tree constructed from BjuA, BnaA, and resequencing of A subgenomes of 17 B. juncea, 5 B. napus and 27 B. rapa accessions. Vegetable- and oil-use subvarieties of B. juncea are marked with black and rose lines. (c) An ultrametric tree constructed from A subgenomes of B. juncea (BjuA) and B. napus (BnaA); two A subgenomes from the resequencing of B. juncea (BjuA_R16) and B. rapa ssp. tricolaris; and two A subgenomes from the resequencing of B. napus (BnaA) and B. rapa ssp. rapa (Europe). B. oleracea (BolC) is considered as an outgroup. Numbers in parentheses indicate the divergence time interval (Mya). The two A subgenomes from the resequencing of B. juncea (BjuA) and B. rapa (ssp. tricolaris) correspond to the two red and bold branches in b. The two A subgenomes from the resequencing of B. napus (BnaA_R1) and B. rapa ssp. rapa (Europe) correspond to the two blue bold branches in b.
Furthermore, we found that all A subgenomes from B. juncea were rooted in the common ancestor, and evolved into different subvarieties for vegetable or oil use (Fig. 2b). Principal component analysis displayed that vegetable- and oil-use subvarieties of B. juncea were distributed nearby B. rapa ssp. tricolaris group and far from other subspecies of B. rapa, supporting the ancestor being closer to B. rapa ssp. tricolaris (Supplementary Fig. 13). Using the independent origin of A subgenomes in B. napus and B. juncea as a control, we compared the SNP variation characteristics between BjuA and BnaA, and that between A subgenomes of vegetable- and oil-use subvarieties of B. juncea. We found typical SNP polyphyletic origin pattern between BjuA and BnaA, and typical SNP monophyletic origin pattern for A subgenomes of vegetable- and oil-use subvarieties in B. juncea (Supplementary Fig. 14). In total, the results drawn from the phylogenetic tree, principal component analysis and SNP variation patterns point to a monophyletic origin and evolution into vegetable- and oil-use subvarieties for A subgenomes of B. juncea.
To estimate when B. juncea formed, we found that the synonymous nucleotide substitution rate was not accurate for estimating formation time of the post-neopolyploid species (Supplementary Fig. 15 and Supplementary Table 26a,b). We therefore used phylogenetic analysis and Bayesian method27 to calculate when BjuA diverged from its closest relative genome (tricolaris; Fig. 2b), to set an upper limit for its time of formation. We considered the time between the divergence of BjuA and the earliest divergent B. juncea accessions (B. juncea; Fig. 2b) as the lower limit for its formation time. We deduced that B. juncea formed ∼0.039−0.055 million years ago (Mya) (Fig. 2c). Similarly, to estimate when B. napus formed, we referred to BnaA and its closest relative genome (European rapa; Fig. 2b) to set an upper limit for its formation time, and to BnaA and the earliest divergent B. napus accessions (B. napus; Fig. 2b) to set a lower limit for its formation time. Here we deduced that B. napus formed 0.038−0.051 Mya (Fig. 2c), which is slightly earlier than the previous estimate of ∼7,500 years ago derived by synonymous substitution (Ks) estimation8.
Homoeolog expression dominance in allopolyploid B. juncea
To explore the transcriptional behavior of the allopolyploid subgenomes, we compared the genome-wide transcriptional levels of homoeologous genes from BjuA and BjuB from different tissues, different developmental stages and two newly resynthesized B. juncea (Supplementary Table 27a). On average, 16.2% of genes displayed homoeolog expression dominance in all samples we investigated, of which we observed only 8.2% to be dominantly expressed towards to BjuB over BjuA excluding resynthesized lines (Fig. 3a and Supplementary Table 27b). This indicated no significant global genome dominance using a double-side binomial test for the subgenomes in B. juncea, which may be explained by the recent polyplodization of this crop. This is consistent with several recent polyploids, such as B. napus8, G. hirsutum17 and T. aestivum19. Transcriptional expression analysis in resynthesized Brassica allopolyploids showed that gene expression changes occurred soon after the initial genome merger and allopolyploidization28. These observations suggest that establishment of homoeolog expression dominance after the initial genome merger and allopolyploidization was immediate. During different developmental stages, 3,339 commonly expressed gene pairs showed homoeolog expression dominance, with 56% of gene pairs displaying dominance toward BjuB subgenomes (Supplementary Fig. 16). In different tissues, 2,251 commonly expressed gene pairs indicated homoeolog expression dominance, and 55% of gene pairs showed dominance toward BjuB (Supplementary Fig. 17). In all evolutionarily synthesized B. juncea, homoeolog expression dominant genes derived predominantly from BjuB, whereas one of the two transcriptomes from the resynthesized B. juncea types showed expression dominance by BjuA (Fig. 3a and Supplementary Table 27b).

(a) Histograms of genome-wide expression of syntenic homoeologous genes among indicated B. juncea tissues and developmental stages. N values indicate the number of dominant genes in BjuA and BjuB, respectively. (b) Boxplot of the distribution of Ka/Ks values between subgenomes (BjuA and BjuB) and among homoeolog expression dominance genes as dominant, subordinate and neutral (non-dominance) in B. juncea. In the left boxplot, Ka/Ks values range from 0.139 to 0.393 with median value 0.238 and interquartile range (IQR) value 0.209 for BjuA, and Ka/Ks values range from 0.147 to 0.411 with median value 0.260 and IQR value 0.224 for BjuB. In the right boxplot, Ka/Ks values range from 0.106 to 0.334 with median value 0.189 and IQR value 0.164 for dominant group, and Ka/Ks values range from 0.193 to 0.438 with median value 0.328 and IQR value 0.279 for subordinate group, and Ka/Ks values range from 0.082 to 0.260 with median value 0.139 and IQR value 0.123 for neutral group. *P < 0.001, permutation test with 1,000 permutations.
We identified 5,632 gene pairs displaying homoeolog expression dominance in B. juncea (Supplementary Table 28). Using the KEGG database, we performed a pathway enrichment analysis for all homoeolog expression dominant genes. This analysis showed that genes showing homoeolog expression dominance were enriched for: cellular processes, environmental information processing, genetic information processing and metabolism and plant-pathogen interaction (Supplementary Fig. 18). Among these pathways, we found that metabolic and plant hormone signal transduction pathways were especially enriched.
We calculated the average non-synonymous/synonymous substitution (Ka/Ks) value of all genes among population accessions based on whole-genome sequencing and resequencing data. The results show significant difference between BjuA and BjuB using a permutation test, of which the BjuB has evolved faster than the BjuA, suggesting asymmetric evolution of the two subgenomes (Fig. 3b). To further analyze homoeolog expression dominant genes, we calculated the average Ka/Ks values among those genes expressed as dominant (higher expression level in homoeologous gene pair), subordinate (lower expression level in homoeologous gene pair) and neutral (equal expression level in homoeologous gene pair). The Ka/Ks values of dominant and subordinate genes (median: 0.31 and 0.35, respectively) were significantly higher than those of neutral genes (median: 0.25) using a permutation test (Fig. 3b). We also calculated the average Ka value among these genes, which indicated the same patterns with Ka/Ks values (Supplementary Fig. 19). This observation indicated that both dominant and subordinate genes evolved more rapidly than did the neutral genes, with subordinate genes being prone to selection in a homoelogous gene pair.
Selection in allopolyploid B. juncea
Using SNPs from resequencing accessions of B. juncea (Supplementary Table 25), we estimated the average pairwise diversity (π) and population differentiation statistics (FST) between the vegetable- and oil-use varieties of B. juncea (Supplementary Tables 29 and 30). We identified selective sweep regions in vegetable- and oil-use B. juncea accessions by combining FST < 0.05 and π ratio < 0.05 outliers (Fig. 4a and Supplementary Table 31). In total, we identified 794 selected genes between the vegetable- and oil-use subvarieties of B. juncea, of which 36.3% (288) showed homoeolog expression dominance. A high proportion of genes with homoeolog expression dominance under selection imply their participation in agricultural trait improvement.

(a) Genome-wide distribution of FST values and π ratio (sliding window = 100 kb, step = 10 kb). Red dashed lines represent the 95% tails for the empirical FST distribution (top) and π ratio (bottom). Magnified are regions of two GSL-metabolism-related genes (BjuA020134 and BjuA020135) and two lipid-metabolism-related genes (BjuA020878 and BjuA030837) that showed both homoeolog expression dominance and strong selective signals, respectively. (b) Sequence analysis of selected genes in a between vegetable- and oil-use subvarieties. The non-synonymous mutation sites are shown on white background. (c) Heat map for genes involved in GSL and lipid synthesis in vegetable-use (highlighted in green) and oil-use (highlighted in orange) subvarieties of B. juncea from the RNA-seq data. Scaled log2 expression values are shown from red to blue in color, indicating high (red) to low (blue) expression. Abbreviations of sample names in c are defined in Supplementary Table 33.
Vegetable-use B. juncea varieties have been selected based on their glucosinolate (GSL) content and composition for human nutrition and plant defense properties29. Oil-use B. juncea varieties have been subjected to intensive breeding to improve their lipid composition, including decreases in the levels of erucic acid and GSL, which are undesirable because they can produce toxic catabolic products in animal feed8. In total, we identified 13 GSL-metabolism-related genes and 22 lipid-metabolism-related genes that were differentially selected between the vegetable- and oil-use subvarieties of B. juncea (Supplementary Table 32). Of these selected genes, 6 GSL-metabolism-related genes and 7 lipid-metabolism-related genes likewise exhibited homoeolog expression dominance (Supplementary Fig. 20 and Supplementary Table 33). One of these genes, BjuB021254, whose ortholog is AT1G04350 (AOP) in Arabidopsis, encodes a 2-oxoglutarate-dependent dioxygenase and has an essential role in GSL biosynthesis30. The gene BjuA030837, whose ortholog is AT1G18460 in Arabidopsis, encodes a lipase family protein that is important in the glycerol biosynthesis process31. The homoeolog expression dominance and selection of these genes suggests that their functions in GSL and lipid metabolism have been subjected to selection and improvement between vegetable and oil-use subvarieties of B. juncea.
With resequencing of vegetable- and oil-use subvarieties of B. juncea, we showed divergent genotypes with nonsynonymous mutation for GSL- and lipid-metabolism-related genes between vegetable- and oil-use subvarieties of B. juncea (Fig. 4b). Oil-use types displayed uniform genotypes for the GSL- and lipid-metabolism-related genes compared to vegetable-use types. In the vegetable-use groups, all but varieties CN59 and CN79 showed consistent genotypes for these genes. We constructed a phylogenetic tree for the vegetable- and oil-use types of B. juncea, in which accessions CN59 and CN79 were clustered into a subgroup independent from the vegetable-use subgroup (Fig. 2b), although we classified them into the vegetable-use subgroup on the basis of their edible organs. Transcriptomic analysis from selected GSL- and lipid-metabolism-associated genes showed significant differences in expression using a two-tailed t-test between vegetable and oil-use subvarieties of B. juncea (Fig. 4c and Supplementary Table 29). These observations indicate that genomic selection has diversified GSL- and lipid-metabolism-related genes between vegetable- and oil-use subvarieties of the plant, each in the direction of their respective agriculturally desirable traits. We also observed 24 selected genes involved in phytohormone metabolism, of which 12 genes exhibited homoeolog expression dominance (Supplementary Table 32). Transcriptomic analysis for selected phytohormone-associated genes showed significant differences in expression using a two tailed t-test between vegetable- and oil-use subvarieties of B. juncea (Supplementary Table 33 and Supplementary Fig. 21). These differences may, likewise, contribute to the phenotypic deviations between the two types.
Discussion
B. rapa, B. juncea and B. napus once comprised the three main Brassica oilseed crops worldwide, whereas at present B. rapa is selected primarily as a vegetable, B. juncea both as a vegetable and for oil use, and B. napus for oilseed32. Discovery of a possible A-subgenome-diversified origin for B. juncea and B. napus may shed light on the unusual features of selection divergence in Brassica. These insights appear promising for de novo synthesis of neo-polyploid species by introgression of individual A-subgenome types to achieve desired breeding purposes.
We demonstrated evidence of homoeolog expression dominance patterns distinguishing A-subgenome types in Brassica. Although homoeolog expression dominance has been observed in several polyploid species17,19,22, a correlation between homoeolog expression dominance and evolutionary rate has not been reported previously. This finding provides important evidence of agricultural selection behavior. More importantly, these observations may facilitate the improvement of agriculturally important traits by focusing selection on the transcriptionally dominant genes.
Selective sweep regions distinguishing vegetable- and oil-use subvarieties of B. juncea identified 794 loci, of which 36.3% showed homoeolog expression dominance. This high proportion of genes with expression dominance under selection implies their potential in agricultural trait improvement by precisely associating homoeolog expression dominance genes with target traits (36.3% selected genes of 16.2% homoeolog expression dominance genes). It is reasonable to assume that this methodology could be applied to a broader array of selected traits in other polyploid crops to provide insight into underlying physiological mechanisms.
Polyploidy is particularly common in flowering plants, is recognized as a characteristic of all angiosperm genomes during their evolution33 and has an essential role in speciation and genomic plasticity34,35. The reprogramming of allopolyploid transcriptomes is shown to be triggered predominantly by interspecific hybridization20, displaying insight into homoeolog gene expression in phenotypic variability and plasticity. Homoeolog expression dominance or bias appears to be a consequence of genome merger and doubling22, but the underlying applicability of the homoeolog expression dominance in agriculture trait selection has not been substantiated to date. We found that homoeolog expression dominant genes have higher Ka/Ks than neutral genes in the allopolyploid B. juncea, consistent with these genes as targets of intensified selection for vegetable- and oil-use varieties of this agriculturally important plant. This observation implies that transcriptional dominance can predate trait selection. The potential linking of homoeolog expression dominance to trait improvement suggests that Brassica breeding programs, and those of other polyploid crops, might benefit from focusing their efforts on the subset of genes with transcriptional dominance, both as a means of enhancing response to selection and toward gaining mechanistic insights.
Methods
Plant materials and sequencing.
Genome sequencing and assembly was done on a B. juncea var. tumida inbred line (T84−66) with excellent agronomic traits being widely used as a parent in breeding (NCBI Biosample SAMN03741772) and a B. nigra double haploid line (YZ12151) (NCBI Biosample SAMN03742614). Sequences of T84−66 included 13 paired-end and mate-paired Illumina libraries (175.8×) and 1 single-molecule reads library (12.03×) combined with 222× of BioNano data (Supplementary Tables 2a,b and 4). Sequences of YZ12151 included 10 paired-end and mate-paired Illumina libraries (95.99×) (Supplementary Table 10). The flow cytometry analysis and the abundance of 17-nt k-mers were performed to estimate the genome size (Supplementary Table 1, and Supplementary Figs. 1 and 5). Additionally, about 10× coverage of genome sequences from 17 B. juncea varieties consisting of 10 vegetable- and 7 oil-use subvarieties for each were generated for genomic analysis (Supplementary Table 24). Low-depth (<1×) genome sequencing of 27 representative B. rapa accessions were used for comparative analysis of A subgenomes in Brassica (Supplementary Note).
De novo assembly.
Genome assembly used ALLPATHS-LG36. All the corrected Pacbio RS II reads were used to fill the gaps by PBjelly_V15.2.20 (ref. 37). RefAligner utility in IrysView was used to perform alignment between Irys molecules and draft assemblies for correcting the scaffolds chimera error. Finally, the corrected scaffolds were anchored to the genomic (optical) maps assembled from BioNano data (Supplementary Fig. 3). This generated assembly v1.0 (Supplementary Table 3). Additional details are available in the Supplementary Note.
Genome quality evaluation.
We used the CEGMA v.2.3 (ref. 25) to blast 458 conserved Core eukaryotic genes (CGE database)24 to assess the genome assembly of B. juncea. The assembled genome of B. juncea was also validated by mapping 23,002 ESTs (length ≥ 500 bp) downloaded from NCBI. To assess the accuracy of the B. juncea genome, we randomly aligned 10 sub-reads over 40 kb from PacBio data to check the paired end relationship using SOAP38 (Supplementary Tables 12 and 14a,b, and Supplementary Fig. 6). Additional details are available in the Supplementary Note.
Genetic map and pseudo-chromosome construction.
We constructed a reference genetic map of B. juncea based on genotyping by resequencing of 100 individuals of F2 population39 (Supplementary Table 5). After resequencing reads alignment with BWA40, potential SNPs were identified by GATK v3.4 (ref. 41). Pairwise recombination of this marker set on each scaffold was calculated, of which adjacent SNPs with pairwise recombination rate less than 0.001 were lumped into a genetic bin, excluding bins showing significantly distorted segregation (chi-squared test, P < 0.01). A final set of bin markers was grouped to 18 linkage groups using Highmap42 (Supplementary Table 8a).
ALLMAPS43 was used to construct the initial pseudo-chromosomes of B. juncea from scaffolds using the genetic map (T84/DTC) constructed in the present study being integrated with a published genetic map (SY/PM)23. We sorted BjuA and BjuB subgenomes of B. juncea referred to the final genetic map (Supplementary Table 9). Additional details are available in the Supplementary Note.
Genome annotation.
The repetitive sequences of the B. juncea genome were identified with a combination of de novo and homolog strategies. Four de novo programs including RepeatScout44, LTR-FINDER45, MITE46 and PILER47 were used to generate the initial repeat library. The initial repeat database was classified into classes, subclasses, superfamilies and families by the PASTEClassifier with REPET48. We then merged transposable element (TE) sequences of Brassica species and the Repbase database49 together to construct a new repeat database and distinguish the genome assembly repeat sequences through RepeatMasker50 (Supplementary Table 15).
Genes were annotated iteratively using three main approaches: homology-based, de novo and EST/unigenes-based. Results of these three methods were integrated by GLEAN51 to get a high-confidence gene model. An RNA-seq based method mapping transcriptome data to the reference genome using TopHat and assembling transcripts with Cufflinks was adopted to obtain the gene structures and new genes52 (Supplementary Tables 18, 19 and 20a,b).
tRNAscan-SEM (version 1.23)53 was used to detect reliable tRNA positions. Noncoding RNAs were predicted by the Infernal program using default parameters54. Through comparing the similarity of secondary structure between the B. juncea sequence and Rfam (v12.0) database55, the noncoding RNAs were classified into different families (Supplementary Table 21).
Stringent criteria and strategy were used to identify new TEs for the BjuA subgenome (Supplementary Fig. 8). The same strategy was used to identify new TEs in the subgenomes of B. juncea and B. napus compared to their corresponding ancestral genome after divergence from a common ancestor (Supplementary Table 16a,b and Supplementary Note).
We performed all-against-all BLASTP (E = 1 × 10−5)56 and chained the BLASTP hits by QUOTA-ALIGN (cscore = 0.5)57 with '1:1 synteny screen' to call synteny blocks. The '1:3 synteny screen' model was used to identify synteny blocks between A. thaliana and Brassica because of whole genome triplication in Brassica evolution history6 by QUOTA-ALIGN (cscore = 0.5). All gene losses were calculated based on the Brassica ancestor common gene sets of each species. Meanwhile, we identified gene loss from other subgenomes (BniB, BjuB, BolC, BnaA and BnaC) of Brassica (Supplementary Tables 22 and 34). Additional details are available in the Supplementary Note.
Comparison of A subgenomes in Brassica.
We called SNPs from A subgenomes by resequencing of B. juncea, B. napus and B. rapa and referring to the B. rapa reference genome using BWA40, GATK41 and SAMtools58 (Supplementary Table 25). Ungenotyped SNPs were imputed by the KNN algorithms59. SNPs with minor allele frequency (MAF) > 0.05 were picked for further analysis. Only non-heterozygous SNPs with integrity > 0.6 were kept for phylogenetic tree construction. The neighbor-joining phylogenetic tree for A subgenomes in Brassica was constructed by MEGA v6.0 using the Kimura 2-parameter model with 1,000 bootstraps and default parameters60.
We selected high quality SNPs with integrity ≥ 0.8 and MAF ≥ 0.05 from all SNPs above for principal component analysis using STRATPCA program from the EIGENSOFT package61.
To compare the characteristics of the SNPs of B. juncea and B. napus, we selected six B. juncea varieties including three vegetable- and three oil-use subvarieties (CN53, CN58, CN04 and CN02, EU07, AU213, respectively) and five B. napus varieties. We only retained SNPs with full integrity (integrity = 1) for further analysis. Fixed SNPs were defined as the frequency of alleles ≥ 60% and were different from their reference genome in B. juncea and B. napus populations. Polymorphic SNPs in B. juncea population were defined as the frequency of alleles ≥ 60%, and their genotypings were distinct from B. napus. Polymorphic SNPs in B. napus population were defined as the frequency of alleles ≥ 60% and their genotypings were dissimilar to B. juncea. We identified fixed and polymorphic SNPs in B. juncea and B. napus populations and those between B. juncea and B. napus population based on different frequencies of alleles scaled at 60%, 70%, 80% and 90%. We used the same strategies to identify fixed and polymorphic SNPs in and between vegetable- and oil-use B. juncea. Additional details are available in the Supplementary Note.
Formation time estimation for B. juncea.
To estimate the formation time of B. juncea, we first selected BjuA, its closest relative genome from B. rapa and the earliest divergent B. juncea accession based on the phylogenetic tree of A subgenomes in Brassica. Then we reconstructed the coding region sequences for selected varieties from the resequencing data. After multiple sequence alignments by MUSCLE v3.3 (ref. 62), a phylogenetic tree was constructed and divergence time was estimated by Bayesian MCMC analyses in BEASTv1.8 (ref. 27) with JIT nucleotide substitution model, relaxed log normal clock model, and one million MCMC generations from which parameters were sampled every 1,000 generations and other default parameters. The divergence time of B. oleracea (4.6 ± 0.5 Mya) was considered as outgroup7. We calculated the divergence time between BjuA and its closest relative genome from B. rapa as the upper limit of formation time of B. juncea. The divergence time between BjuA and the earliest divergent B. juncea accessions was considered as the lower limit of formation time of B. juncea. Additional details are available in the Supplementary Note.
Homoeolog expression dominance analysis.
The clean reads from RNA-seq after quality control were mapped onto the B. juncea genome using Tophat2 (ref. 63). The gene expression level of individual genes was quantified using RPKM values (fragments per kilobase of exon per million fragments mapped) by Cufflinks52. Homoeolog expression dominance analysis was performed within syntenic gene pairs. Differentially expressed genes pairs with greater than twofold change were defined as dominant gene pairs. The dominant genes were the genes that were expressed relatively higher in dominant gene pairs, and the lower ones are the subordinate genes. The rest of the syntenic gene pairs that showed non-dominance were classified as neutral genes. To test whether the occurrences of BjuA dominant gene pairs and the occurrences of BjuB dominant gene pairs are equal, we performed double-side binomial tests on dominant gene pairs for all samples64 (Supplementary Table 27a,b). Additional details are available in the Supplementary Note.
Selective pressure on dominantly expressed genes and subgenomes.
All SNP sets were called by GATK41 for 17 B. juncea accessions with default parameters and filtered out with depth < 3×. Coding region sequence sets were then reconstructed based on high quality SNPs for each sample. To detect selective pressure of each coding gene, the rates of nonsynonymous (dN) and synonymous (dS) (ω = dN/dS) substitutions were estimated site-by-site using the YN00 program with default parameters from the PAML 4.2b package58. Each paired gene set of 17 samples was estimated repeatedly. All Ka/Ks of gene pairs were classified to three categories (dominant genes, subordinate genes and neutral genes). Meanwhile All Ka/Ks of gene pairs were separated into BjuA/BjuB subgenomes. To test statistical significance of different data sets, we performed a permutation test on them with 1,000 permutations (Supplementary Table 26a,b). Additional details are available in the Supplementary Note.
Detection of selective sweep signals.
Average pairwise diversity (π) and population differentiation statistic (FST) were calculated through Bio::PopGen of bioperl package65. Selective sweep regions were identified in the 10 vegetable- and 7 oil-use B. juncea subvarieties by combining FST outliers and π ratio outliers (θπ (vegetable-use/oil-use)) with 100 kb sliding windows and 10 kb steps. Adjacent windows extended to 10 kb likely represent the effect of a single divergence region and thus were linked to define a 'candidate gene region' (Supplementary Table 31). Additional details are available in the Supplementary Note.
URLs.
PBjelly, https://sourceforge.net/projects/pb-jelly/; A. thaliana and B. rapa protein sequences, http://genome.jgi-psf.org/; PASA, http://pasapipeline.github.io/; KEGG Automatic Annotation Server; http://www.genome.jp/kegg/kaas/.
Accession codes.
The genome assemblies of B. juncea and B. nigra have been deposited at GenBank: LFQT00000000 and LFLV00000000, respectively (BioProject PRJNA285130).
Accession codes
Change history
24 September 2018
Following publication of this article, the authors have corrected 426 chimeric scaffolds in this genome (total scaffold number 10,684). The genome assembly has now been improved as V1.5, and the updated genome assembly is available to be downloaded from http://brassicadb.org/brad/datasets/pub/Genomes/Brassica_juncea/V1.5/.
References
Warwick, S.I., Francis, A. & Al-Shehbaz, I.A. Brassicaceae: species checklist and database on CD–Rom. Plant Syst. Evol. 259, 249–258 (2006).
Nagaharu, U. Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilication. Jpn. J. Bot. 7, 389–452 (1935).
Chen, S. et al. Evidence from genome-wide simple sequence repeat markers for a polyphyletic origin and secondary centers of genetic diversity of Brassica juncea in China and India. J. Hered. 104, 416–427 (2013).
Institute of Archaeology of Chinese Academy of Science. Xian Banpo country. Special Issue of Archaeology (Archaeology Press, 1963).
Prakash, S. & Hinata, K. Taxonomy, cytogenetics and origin of crop Brassicas, a review. Opera Bot. 55, 1–57 (1980).
Wang, X. et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–1039 (2011).
Liu, S. et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 5, 3930 (2014).
Chalhoub, B. et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345, 950–953 (2014).
Lysak, M.A., Koch, M.A., Pecinka, A. & Schubert, I. Chromosome triplication found across the tribe Brassiceae. Genome Res. 15, 516–525 (2005).
Bowers, J.E., Chapman, B.A., Rong, J. & Paterson, A.H. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 422, 433–438 (2003).
Cheng, F. et al. Deciphering the diploid ancestral genome of the mesohexaploid Brassica rapa. Plant Cell 25, 1541–1554 (2013).
Town, C.D. et al. Comparative genomics of Brassica oleracea and Arabidopsis thaliana reveal gene loss, fragmentation, and dispersal after polyploidy. Plant Cell 18, 1348–1359 (2006).
Mun, J.H. et al. Genome-wide comparative analysis of the Brassica rapa gene space reveals genome shrinkage and differential loss of duplicated genes after whole genome triplication. Genome Biol. 10, R111 (2009).
Michael, T.P. & VanBuren, R. Progress, challenges and the future of crop genomes. Curr. Opin. Plant Biol. 24, 71–81 (2015).
Adams, K.L., Cronn, R., Percifield, R. & Wendel, J.F. Genes duplicated by polyploidy show unequal contributions to the transcriptome and organ-specific reciprocal silencing. Proc. Natl. Acad. Sci. USA 100, 4649–4654 (2003).
Flagel, L., Udall, J., Nettleton, D. & Wendel, J. Duplicate gene expression in allopolyploid Gossypium reveals two temporally distinct phases of expression evolution. BMC Biol. 6, 16 (2008).
Zhang, T. et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 33, 531–537 (2015).
Bottley, A., Xia, G.M. & Koebner, R.M. Homoeologous gene silencing in hexaploid wheat. Plant J. 47, 897–906 (2006).
International Wheat Genome Sequencing Consortium (IWGSC). A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345, 1251788 (2014).
Chang, P.L., Dilkes, B.P., McMahon, M., Comai, L. & Nuzhdin, S.V. Homoeolog-specific retention and use in allotetraploid Arabidopsis suecica depends on parent of origin and network partners. Genome Biol. 11, R125 (2010).
Wang, J. et al. Stochastic and epigenetic changes of gene expression in Arabidopsis polyploids. Genetics 167, 1961–1973 (2004).
Grover, C.E. et al. Homoeolog expression bias and expression level dominance in allopolyploids. New Phytol. 196, 966–971 (2012).
Ostergaard, L. & King, G.J. Standardized gene nomenclature for the Brassica genus. Plant Methods 4, 10 (2008).
Zou, J. et al. Co-linearity and divergence of the A subgenome of Brassica juncea compared with other Brassica species carrying different A subgenomes. BMC Genomics 17, 18 (2016).
Ye, Y.N., Hua, Z.G., Huang, J., Rao, N. & Guo, F.B. CEG: a database of essential gene clusters. BMC Genomics 14, 769 (2013).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Drummond, A.J., Suchard, M.A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973 (2012).
Zhang, D. et al. Genome-specific differential gene expressions in resynthesized Brassica allotetraploids from pair-wise crosses of three cultivated diploids revealed by RNA-seq. Front. Plant Sci. 6, 957 (2015).
Verkerk, R. et al. Glucosinolates in Brassica vegetables: the influence of the food supply chain on intake, bioavailability and human health. Mol. Nutr. Food Res. 53 (Suppl. 2), S219–S265 (2009).
Sønderby, I.E., Geu-Flores, F. & Halkier, B.A. Biosynthesis of glucosinolates-gene discovery and beyond. Trends Plant Sci. 15, 283–290 (2010).
Li-Beisson, Y. et al. Acly-lipid metabolism. in The Arabidopsis Book. The American Society of Plant Biologists 8, e0133 (2010).
Schmidt, R. & Bancroft, I. Brassicaceae in agriculture. in Genetics and Genomics of the Brassicaceae 33–65 (Springer, 2011).
Jiao, Y. et al. Ancestral polyploidy in seed plants and angiosperms. Nature 473, 97–100 (2011).
Leitch, A.R. & Leitch, I.J. Genomic plasticity and the diversity of polyploid plants. Science 320, 481–483 (2008).
Soltis, P.S. & Soltis, D.E. The role of hybridization in plant speciation. Annu. Rev. Plant Biol. 60, 561–588 (2009).
Maccallum, I. et al. ALLPATHS 2: small genomes assembled accurately and with high continuity from short paired reads. Genome Biol. 10, R103 (2009).
English, A.C. et al. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS One 7, e47768 (2012).
Gu, S., Fang, L. & Xu, X. Using SOAPaligner for short reads alignment. Curr. Protoc. Bioinformatics 44, 1–17 (2013).
Huang, X. et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 19, 1068–1076 (2009).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
DePristo, M.A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Liu, D. et al. Construction and analysis of high-density linkage map using high-throughput sequencing data. PLoS One 9, e98855 (2014).
Tang, H. et al. ALLMAPS: robust scaffold ordering based on multiple maps. Genome Biol. 16, 3 (2015).
Price, A.L., Jones, N.C. & Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 21 (Suppl. 1), i351–i358 (2005).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Han, Y. & Wessler, S.R. MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 38, e199 (2010).
Edgar, R.C. & Myers, E.W. PILER: identification and classification of genomic repeats. Bioinformatics 21 (Suppl. 1), i152–i158 (2005).
Wicker, T., Matthews, D.E. & Keller, B. TREP: a database for Triticeae repetitive elements. Trends Plant Sci. 7, 561–562 (2002).
Bao, W., Kojima, K.K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11 (2015).
Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics Chapter 4, Unit 4.10 (2004).
Elsik, C.G. et al. Creating a honey bee consensus gene set. Genome Biol. 8, R13 (2007).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Lowe, T.M. & Eddy, S.R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Nawrocki, E.P. & Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Nawrocki, E.P. et al. Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 43, D130–D137 (2015).
Kiełbasa, S.M., Wan, R., Sato, K., Horton, P. & Frith, M.C. Adaptive seeds tame genomic sequence comparison. Genome Res. 21, 487–493 (2011).
Tang, H. et al. Screening synteny blocks in pairwise genome comparisons through integer programming. BMC Bioinformatics 12, 102 (2011).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Chen, W. et al. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat. Genet. 46, 714–721 (2014).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Price, A.L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006).
Edgar, R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Schnable, J.C., Springer, N.M. & Freeling, M. Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc. Natl. Acad. Sci. USA 108, 4069–4074 (2011).
Stajich, J.E. et al. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 12, 1611–1618 (2002).
Acknowledgements
We thank X.H. Qi for the construction of F2 population for genetic mapping, and L.J. Fan for the flow cytometry analysis. This work was supported by grants from Science and Technology Program of Zhejiang Province (2015C32046), Ear-marked Special Fund from Ministry of Agriculture (09162130135252) and the National Natural Science Foundation of China (31372063).
Author information
Authors and Affiliations
Contributions
J.Y., M.Z. and S.A.M. designed the project. D.L., X.W., C.J., F.C., B.L., Y.J., P.Y., X.L., K.X., H.Z., J.Z. and J.Y. performed most genome sequencing and bioinformatics analyses. S.C., J.W. and F.L. contributed some oil-use B. juncea and B. nigra varieties and discussed the project. D.P. donated B. juncea BAC sequences. Z.H., W.M. and J.S. prepared F2 population materials for genetic mapping and DNA extraction. J.Y., M.Z. and C.J. wrote the manuscript. S.A.M. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–21, Supplementary Tables 1–4, 6–14, 16–27, 31 and 34, and Supplementary Note. (PDF 2929 kb)
Supplementary Table 5
Resequencing of B. juncea F2 population for genetic map construction. (XLSX 17 kb)
Supplementary Table 15
Summary of repeat content in Brassica rapa, Brassica nigra and Brassica oleracea. (XLSX 16 kb)
Supplementary Table 28
The homoelog expression dominance genes list of B. juncea from mixc. (XLSX 377 kb)
Supplementary Table 29
List of regions associated with FST (significance < 0.05). (XLSX 242 kb)
Supplementary Table 30
List of regions associated with Π ratio (significance < 0.05). (XLSX 263 kb)
Supplementary Table 32
Function annotation of GSLs and lipid-related genes from FST and π in B. juncea. (XLSX 18 kb)
Supplementary Table 33
Expression level of glucosinolate, lipid, auxin and ethylene related genes in different tissues of B. juncea. (XLS 82 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International licence. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, J., Liu, D., Wang, X. et al. The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat Genet 48, 1225–1232 (2016). https://doi.org/10.1038/ng.3657
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3657
This article is cited by
-
Genetic manipulation of Indian mustard genotypes with WRR-gene(s) confers resistance against Albugo candida
Molecular Biology Reports (2024)
-
Differential subgenome expression underlies biomass accumulation in allotetraploid Pennisetum giganteum
BMC Biology (2023)
-
Analyses of a chromosome-scale genome assembly reveal the origin and evolution of cultivated chrysanthemum
Nature Communications (2023)
-
The allotetraploid horseradish genome provides insights into subgenome diversification and formation of critical traits
Nature Communications (2023)
-
Genetic mapping of some key plant architecture traits in Brassica juncea using a doubled haploid population derived from a cross between two distinct lines: vegetable type Tumida and oleiferous Varuna
Theoretical and Applied Genetics (2023)
