
Abstract
There is increasing evidence supporting the role of genetic variants in the development of radiation-induced toxicity1. However, previous candidate gene association studies failed to elucidate the common genetic variation underlying this phenotype2, which could emerge years after the completion of treatment3. We performed a genome-wide association study on a Spanish cohort of 741 individuals with prostate cancer treated with external beam radiotherapy (EBRT). The replication cohorts consisted of 633 cases from the UK4 and 368 cases from North America5. One locus comprising TANC1 (lowest unadjusted P value for overall late toxicity = 6.85 × 10−9, odds ratio (OR) = 6.61, 95% confidence interval (CI) = 2.23–19.63) was replicated in the second stage (lowest unadjusted P value for overall late toxicity = 2.08 × 10−4, OR = 6.17, 95% CI = 2.25–16.95; Pcombined = 4.16 × 10−10). The inclusion of the third cohort gave unadjusted Pcombined = 4.64 × 10−11. These results, together with the role of TANC1 in regenerating damaged muscle, suggest that the TANC1 locus influences the development of late radiation-induced damage.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout

Similar content being viewed by others

Accession codes
References
West, C.M. & Barnett, G.C. Genetics and genomics of radiotherapy toxicity: towards prediction. Genome Med. 3, 52 (2011).
Andreassen, C.N. Searching for genetic determinants of normal tissue radiosensitivity—are we on the right track? Radiother. Oncol. 97, 1–8 (2010).
Bentzen, S.M. et al. Normal tissue effects: reporting and analysis. Semin. Radiat. Oncol. 13, 189–202 (2003).
Burnet, N.G. et al. RAPPER: the radiogenomics of radiation toxicity. Clin. Oncol. (R. Coll. Radiol.) 25, 431–434 (2013).
Kerns, S.L. et al. Genome-wide association study to identify single nucleotide polymorphisms (SNPs) associated with the development of erectile dysfunction in African-American men after radiotherapy for prostate cancer. Int. J. Radiat. Oncol. Biol. Phys. 78, 1292–1300 (2010).
Jemal, A. et al. Cancer statistics, 2008. CA Cancer J. Clin. 58, 71–96 (2008).
Matzinger, O. et al. Acute toxicity of curative radiotherapy for intermediate- and high-risk localised prostate cancer in the EORTC trial 22991. Eur. J. Cancer 45, 2825–2834 (2009).
Lawton, C.A. et al. Long-term treatment sequelae following external beam irradiation for adenocarcinoma of the prostate: analysis of RTOG studies 7506 and 7706. Int. J. Radiat. Oncol. Biol. Phys. 21, 935–939 (1991).
Barnett, G.C. et al. Independent validation of genes and polymorphisms reported to be associated with radiation toxicity: a prospective analysis study. Lancet Oncol. 13, 65–77 (2012).
Barnett, G.C. et al. Individual patient data meta-analysis shows no association between the SNP rs1800469 in TGFB and late radiotherapy toxicity. Radiother. Oncol. 105, 289–295 (2012).
Andreassen, C.N. et al. Conducting radiogenomic research—do not forget careful consideration of the clinical data. Radiother. Oncol. 105, 337–340 (2012).
Barnett, G.C. et al. The impact of clinical factors on the development of late radiation toxicity: results from the medical research council RT01 trial (ISRCTN47772397). Clin. Oncol. (R. Coll. Radiol.) 23, 613–624 (2011).
Kerns, S.L. et al. A 2-stage genome-wide association study to identify single nucleotide polymorphisms associated with development of erectile dysfunction following radiation therapy for prostate cancer. Int. J. Radiat. Oncol. Biol. Phys. 85, e21–e28 (2013).
Kerns, S.L. et al. A two-stage genome-wide association study to identify single nucleotide polymorphisms associated with development of urinary symptoms following radiotherapy for prostate cancer. J. Urol. 190, 102–108 (2013).
Kerns, S.L. et al. Genome-wide association study identifies a region on chromosome 11q14.3 associated with rectal bleeding following radiation therapy for prostate cancer. Radiother. Oncol. 107, 372–376 (2013).
Barnett, G.C. et al. A genome wide association study (GWAS) providing evidence of an association between common genetic variants and late radiotherapy toxicity. Radiother. Oncol. 10.1016/j.radonc.2014.02.012 (28 April 2014).
Kerns, S.L. et al. STROGAR—STrengthening the Reporting Of Genetic Association studies in Radiogenomics. Radiother. Oncol. 110, 182–188 (2014).
Howie, B.N., Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 5, e1000529 (2009).
Howie, B., Marchini, J. & Stephens, M. Genotype imputation with thousands of genomes. G3 1, 457–470 (2011).
1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012).
Li, Y., Willer, C., Sanna, S. & Abecasis, G. Genotype imputation. Annu. Rev. Genomics Hum. Genet. 10, 387–406 (2009).
Barnett, G.C. et al. Standardized Total Average Toxicity Score: a scale- and grade-independent measure of late radiotherapy toxicity to facilitate pooling of data from different studies. Int. J. Radiat. Oncol. Biol. Phys. 82, 1065–1074 (2012).
Bentzen, S.M. & Overgaard, J. Patient-to-patient variability in the expression of radiation-induced normal tissue injury. Semin. Radiat. Oncol. 4, 68–80 (1994).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Gabriel, S.B. et al. The structure of haplotype blocks in the human genome. Science 296, 2225–2229 (2002).
ENCODE Project Consortium. A user's guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 9, e1001046 (2011).
Yang, T.P. et al. Genevar: a database and Java application for the analysis and visualization of SNP-gene associations in eQTL studies. Bioinformatics 26, 2474–2476 (2010).
Stranger, B.E. et al. Patterns of cis regulatory variation in diverse human populations. PLoS Genet. 8, e1002639 (2012).
Rau, A. et al. rolling pebbles (rols) is required in Drosophila muscle precursors for recruitment of myoblasts for fusion. Development 128, 5061–5073 (2001).
Avirneni-Vadlamudi, U. et al. Drosophila and mammalian models uncover a role for the myoblast fusion gene TANC1 in rhabdomyosarcoma. J. Clin. Invest. 122, 403–407 (2012).
Abmayr, S.M. & Pavlath, G.K. Myoblast fusion: lessons from flies and mice. Development 139, 641–656 (2012).
Edwards, S.L., Beesley, J., French, J.D. & Dunning, A.M. Post-GWAS studies: illuminating the dark road from association to function. Am. J. Hum. Genet. 93, 779–797 (2013).
West, C. et al. Establishment of a Radiogenomics Consortium. Int. J. Radiat. Oncol. Biol. Phys. 76, 1295–1296 (2010).
Talbot, C.J. et al. A replicated association between polymorphisms near TNFα and risk for adverse reactions to radiotherapy. Br. J. Cancer 107, 748–753 (2012).
Fachal, L. et al. Association of a XRCC3 polymorphism and rectum mean dose with the risk of acute radio-induced gastrointestinal toxicity in prostate cancer patients. Radiother. Oncol. 105, 321–328 (2012).
Fachal, L. et al. TGFβ1 SNPs and radio-induced toxicity in prostate cancer patients. Radiother. Oncol. 103, 206–209 (2012).
Dearnaley, D.P. et al. Escalated-dose versus standard-dose conformal radiotherapy in prostate cancer: first results from the MRC RT01 randomised controlled trial. Lancet Oncol. 8, 475–487 (2007).
Dearnaley, D. et al. Conventional versus hypofractionated high-dose intensity-modulated radiotherapy for prostate cancer: preliminary safety results from the CHHiP randomised controlled trial. Lancet Oncol. 13, 43–54 (2012).
Vogelius, I.R. & Betzen, S.M. Meta-analysis of the α/β ratio for prostate cancer in the presence of an overall time factor: bad news, good news, or no news? Int. J. Radiat. Oncol. Biol. Phys. 85, 89–94 (2013).
Amigo, J., Salas, A., Costas, J. & Carracedo, A. GDF: dealing with high-throughput genotyping multiplatform data for medical and population genetic applications. J. Proteomics Bioinform. 5, 1–6 (2012).
Delaneau, O., Marchini, J. & Zagury, J.-F. A linear complexity phasing method for thousands of genomes. Nat. Methods 9, 179–181 (2012).
Skol, A.D., Scott, L.J., Abecasis, G.R. & Boehnke, M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat. Genet. 38, 209–213 (2006).
Barrett, J.C., Fry, B., Maller, J. & Daly, M.J. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21, 263–265 (2005).
Acknowledgements
We are grateful to all participating patients for their cooperation. We thank I. Quintela from CEGEN (Spanish National Genotyping Center) for her support as Affymetrix genotyping platform manager. We also thank the Galician Supercomputation Centre for providing computing infrastructures and the Department of Scientific Computing at the Icahn School of Medicine at Mount Sinai for providing computational resources and staff expertise. This work was funded by grants from the Instituto de Salud Carlos III (FIS PI10/00164 and PI13/02030) and the Fondo Europeo de Desarrollo Regional (FEDER 2007–2013). L.F. is supported by the Isabel Barreto program from Xunta de Galicia and Fondo Social Europeo and was granted by an ESTRO Technology Transfer Grant (2012). The RAPPER group is supported by Cancer Research UK and Experimental Cancer Medicine Centre funding. Investigators from the Institute of Cancer Research and the Royal Marsden NHS Foundation Trust acknowledge support from the National Institute for Health Research (NIHR) Royal Marsden NHS Foundation Trust and The Institute of Cancer Research Biomedical Research Centre. Sample and data collection within the CHHiP trial (CRUK/06/16) were supported by Cancer Research UK (SP2312/021 and C8262/A7253) and the UK Department of Health, with patient recruitment at participating sites supported by the NIHR Cancer Research Network. B.S.R. and S.L.K. are funded by grants RSGT-05-200-01-CCE from the American Cancer Society, PC074201 from the US Department of Defense and 1R01CA134444 from the US National Institutes of Health. Á.C. acknowledges support from the Instituto de Salud Carlos III and the Fondo Europeo de Desarrollo Regional (PI13/01136, FEDER 2007–2013), a King Abdulaziz University grant (1-117-1434-HiCi), Innopharma and the Botin Foundation. The research collaboration leading to this paper was developed under the framework of the Radiogenomics Consortium.
Author information
Authors and Affiliations
Contributions
A.V. conceived and coordinated the study and obtained financial support. A.V. and L.F. designed the GWAS. L.F. performed laboratory work, data analysis, quality control analysis and imputation of the RADIOGEN study. L.F. and A.V. drafted the manuscript with contributions from C.M.L.W., G.C.B., A.M.D., S.L.K., B.S.R. and L.D. L.F., A.G.-C. and A.V. collated and managed the RADIOGEN database. A.G.-C., P.P., A.M.C. and P.C.-C. were involved in the collection of cases, and clinical and toxicity data for the RADIOGEN study. G.C.B., N.G.B., C.M.L.W. and A.M.D. were involved in the conception and design of the RAPPER study. G.C.B. and L.D. were involved in quality control analysis and imputation for the RAPPER study. G.C.B., R.M.E., D.P.D., M.R.S. and E.H. were involved in RAPPER data acquisition. S.L.K. was involved in laboratory work, quality control analysis and imputation for the Gene-PARE study. M.S.-G. and R.L.-B. were involved in the collection of dosimetric data for the RADIOGEN study. D.P.D. designed, initiated and was chief investigator for the RAPPER RT01 and CHHiP trials. Á.C. contributed to the RADIOGEN study. B.S.R. was involved in the conception and design of the Gene-PARE study. All authors were involved in the writing or critical review of the draft report, and all approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
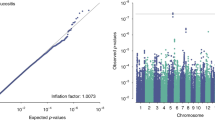
Supplementary Figure 2 Manhattan plot of observed P values from univariate analysis.
(a) STATacute and (b) STATlate scores in the RADIOGEN cohort. The blue line delimits the P-value threshold of 1 × 10−5.
Supplementary Figure 3 QQ plots of P-value distribution from the RADIOGEN univariate analysis.
A: STATacute; B: STATlate.
Supplementary Figure 4 Haploview analyses of the TANC1 locus.
The linkage disequilibrium color scheme corresponds with the D′ parameter, while linkage disequilibrium values correspond to the r2 parameter. (a) Haploview analysis in the RADIOGEN cohort. The red box highlights the block where the most significant SNPs are placed. The left bottom diagram shows the linkage disequilibrium for the HapMap CEU and TSI population at the same region. (b) Haploview analysis of the replicated SNPs in the three cohorts (RADIOGEN, RAPPER and Gene-PARE).
Supplementary Figure 5 ENCODE data for the TANC1 locus.
(a) TANC1 coding region. (b) Expanded view of the LD block containing all the associated variants. (c) Expanded view of the site where the responsible SNPs are located, after conditional analyses, of all the identified effect in the RADIOGEN cohort (rs264663). The UCSC Gene annotation track is a set of gene predictions based on data from RefSeq, GenBank, CCDS, Rfam and the tRNA Genes track. (Data last updated 14 June 2013.) The Chromatin State Segmentation track displays a chromatin state segmentation for each of nine human cell types.. The H3K4me3 track shows the levels of enrichment of the H3K4me3 histone mark across the genome as determined by a ChIP-seq assay. The H3K4me3 histone mark is the trimethylation of lysine 4 of the H3 histone protein, and it is associated with promoters that are active or poised to be activated. The H3K4me1 track shows the levels of enrichment of the H3K4me1 histone mark across the genome as determined by a ChIP-seq assay. The H3K4me1 histone mark is the monomethylation of lysine 4 of the H3 histone protein, and it is associated with enhancers and with DNA regions downstream of transcription start sites. The H3K27ac track shows the levels of enrichment of the H3K27ac histone mark across the genome as determined by a ChIP-seq assay. The H3K27ac histone mark is the acetylation of lysine 27 of the H3 histone protein, and it is thought to enhance transcription, possibly by blocking the spread of the repressive histone mark H3K27me3. The DNase clusters track shows DNase hypersensitive areas. Tnx Factor track shows regions where transcription factors, proteins responsible for modulating gene transcription, bind to DNA, as assayed by ChIP-seq.
Supplementary Figure 6 eQTL analyses performed in the TANC1 locus with Genevar in HapMap populations.
(a) Gene-centric analysis results. Observed eQTL SNPs (eSNPs) are visualized as a regional plot (1 Mb), where a dotted line represents the P-value threshold (0.01). The blue box highlights where rs264663 is located. rs264663 does not reach the MAF threshold of 0.05 in the CEU populations required by Genevar to be analyzed. Note, however, that the SNP is found in the locus containing the eSNPs with the lowest P values. (b) Best candidate eSNPs after runing 10,000 permutations identified by gene-centric analysis. The red box highlights rs264663. ρ, Spearman's rank correlation coefficient; P, P value; Pemp, adjusted P value after running 10,000 permutations. (c) SNP-gene association analysis results. (d,e) SNP-centric analysis results. Note that positions refer to assembly NCI36/hg18. According to this assembly, TANC1 is located between coordinates 159,533,392 and 159,797,412.
Supplementary Figure 7 Plot of STATlate against the biological effective dose in the RADIOGEN cohort.
Lines represent linear regression results from STATlate = β0 + β × BED + E, where β is represented by the slope of the lines. rs264663 CC β = 7.61 × 10−4; rs264663 CT β = 0.03. The regression model for the RADIOGEN cohort (N = 410; BED could not be estimated in 7 of the 417 patients with late toxicity report) is represented by the blue line. The black line represents the regression model for individuals who do not harbor the rs26466 risk allele (N = 395), whereas the red line represent the regression model for patients harboring a risk allele (N = 15). Note that this discrepancy in the slope between the black and red lines is what the statistical interaction in Supplementary Table 7 is reflecting.
Supplementary Figure 8 Histograms of standardized total average toxicity (STAT) scores and residuals (R-STAT).
(a−e) Frequency distributions of STAT score on the RADIOGEN (a,b), RAPPER (c,d) and Gene-PARE (e) cohorts. (f−i) Frequency distributions of R-STAT score on the RADIOGEN (f,g) and RAPPER (h,i) cohorts, which approximately follow a normal distribution. There are no R-STAT data for the Gene-PARE cohort because no associated covariates were identified in this cohort. Light blue refers to overall acute toxicity, while dark blue refers to overall late toxicity. The dashed line shows the threshold of 1 s.d. after the median used to dichotomize the variable.
Supplementary Figure 9 Principal-component analyses in the RADIOGEN cohort.
(a) Ancestry PCA in RADIOGEN. Black, green and red circles represent HapMap samples (HapMap r23a) of European, Asian and African descent, respectively. Blue circles represent RADIOGEN samples. One sample was excluded after ancestry PCA analysis. (b) PCA performed including only RADIOGEN samples beyond the boundaries of the main cluster (PC1 < −50, PC2 > 50 < −50) were removed from the analysis. Sixty-eight samples were excluded after this second PCA analysis.
Supplementary Figure 10 Example of visual inspection of cluster plots for SNPs that demonstrated evidence of association in the first stage (RADIOGEN cohort).
SNP cluster priors (red, blue and green dashed-line cluster boundaries as defined by Affymetrix training data indicating the prior localization of the genotype clusters) and SNP cluster posteriors (solid-line cluster boundaries as defined by experimental data combining information from priors) are shown. The gray and pink dashed line indicates the localization (log radio, x axis) of the prior for each allele (noted by Affymetrix as A or B). The final genotype for each sample is indicated by red triangles (homozygous for allele A), blue squares (heterozygous samples) and green circles (homozygous for allele B).
Supplementary Figure 11 Principal-component analysis in the RAPPER cohort.
Black triangles indicate the ethnicity of patients in the RAPPER study. Blue, green and red circles represent HapMap samples of European, Asian and African descent, respectively. Blue circles are nearly obscured by the black triangles. The majority of RAPPER subjects cluster with HapMap Europeans and largely obscure the blue circles. Twenty-two prostate cancer patients with non-Caucasian ancestry were excluded (see Barnet et al.16).
Supplementary Figure 12 Principal-component analysis in the Gene-PARE cohort.
(a) HapMap samples showing the distribution of 11 reference populations. HapMap samples are shown by colored circles, and Gen-PARE patients are shown by black/gray crosses. YRI, Yoruban in Ibadan, Nigeria; LWK, Luhya in Webuye, Kenya; MKK, Maasai in Kinyawa, Kenya; ASW; African ancestry in Southwest USA; CEU, Utah residents with Northern and Western European ancestry; TSI, Tuscan in Italy; GIH, Gujarati Indians in Houston, Texas; MEX, Mexican ancestry in Los Angeles, California; CHB, Han Chinese in Beijing, China; CHD, Chinese in Metropolitan Denver, Colorado; JPT, Japanese in Tokyo, Japan (see Kerns et al.13). (b) Samples beyond the boundaries of the main cluster (PC1 < 1, PC2 > 1) were removed from the analysis in the present study.
Supplementary information
Supplementary Text and Figures
Supplementary Tables 1–16, Supplementary Figures 1–12 and Supplementary Note (PDF 4377 kb)
Rights and permissions
About this article

Cite this article
Fachal, L., Gómez-Caamaño, A., Barnett, G. et al. A three-stage genome-wide association study identifies a susceptibility locus for late radiotherapy toxicity at 2q24.1. Nat Genet 46, 891–894 (2014). https://doi.org/10.1038/ng.3020
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3020
This article is cited by
-
Effect of genetic polymorphisms on outcomes following nivolumab for advanced renal cell carcinoma in the SNiP-RCC trial
Cancer Immunology, Immunotherapy (2023)
-
A two-stage genome-wide association study to identify novel genetic loci associated with acute radiotherapy toxicity in nasopharyngeal carcinoma
Molecular Cancer (2022)
-
TANC1 methylation as a novel biomarker for the diagnosis of patients with anti-tuberculosis drug-induced liver injury
Scientific Reports (2021)
-
Biobanking in radiotherapy trials — a challenge to the clinical research community
Nature Reviews Clinical Oncology (2021)
-
Genetically-regulated transcriptomics & copy number variation of proctitis points to altered mitochondrial and DNA repair mechanisms in individuals of European ancestry
BMC Cancer (2020)
