
Abstract
The initiation of flowering in plants is controlled by environmental and endogenous signals1,2. Molecular analysis of this process in Arabidopsis thaliana indicates that environmental control is exerted through the photoperiod and vernalization pathways, whereas endogenous signals regulate the autonomous and gibberellin pathways. The vernalization and autonomous pathways converge on the negative regulation of FLC3,4, a gene encoding a MADS-box protein that inhibits flowering3,4. We cloned FVE, a component of the autonomous pathway that encodes AtMSI4, a putative retinoblastoma-associated protein. FVE interacted with retinoblastoma protein in immunoprecipitation assays, and FLC chromatin was enriched in acetylated histones in fve mutants. We conclude that FVE participates in a protein complex repressing FLC transcription through a histone deacetylation mechanism. Our data provide genetic evidence of a new developmental function of these conserved proteins and identify a new genetic mechanism in the regulation of flowering.
Similar content being viewed by others


Main
Many plants require prolonged exposure to near-freezing temperatures (vernalization) to flower. This reversible delay is associated in A. thaliana with the expression of the FLC gene3,4. In early flowering plants that do not require vernalization, FLC is expressed at low levels because these plants lack a functional allele at the FRI locus that encodes an FLC activator5. Genes of the flowering autonomous pathway, such as FCA, FPA, FVE, FY, LD and FLD also restrict FLC expression in early-flowering plants6,7. Mutations in these genes increase levels of FLC mRNA, resulting in a photoperiod-independent flowering delay. The increased FLC expression and late flowering of plants with mutations of the autonomous pathway can be reverted by vernalization3,4, and flc null mutations completely correct the late flowering of plants with mutations of this pathway8. These effects indicate that the primary function of the autonomous pathway is the negative regulation of FLC. Thus, FLC integrates flowering signals from the vernalization and autonomous pathways. Molecular identification of several genes in the autonomous pathway suggested potential roles as regulators of FLC expression based on the function assigned to the deduced proteins9,10,11,12. But this information has not provided an understanding of the molecular mechanisms involved in the regulation of FLC expression.
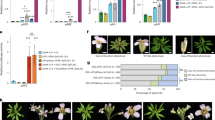
FVE is a classical flowering time locus of the autonomous pathway6,13,14, as fve mutants flowered later than wild-type plants in any photoperiod condition (Fig. 1). The delay in flowering time caused by the fve mutations was completely corrected by exposure to 4 °C for 12 weeks (Fig. 1b). In addition, fve mutations caused an increase in the steady state level of the FLC transcript (Fig. 1c), which can be reverted by vernalization3,4. For further insight into the regulation of flowering time by the autonomous pathway, we cloned the gene FVE with a map-based approach (Supplementary Fig. 1 online). A genomic sequence on chromosome 2 previously annotated as AtMSI4 (ref. 15) was identified as FVE because it carried mutations in four fve alleles (Fig. 1d). In addition, fve-1 and fve-3 plants were complemented with a FVE genomic fragment (Fig. 2a). FVE encodes a predicted protein of 507 amino acids with six WD repeat domains that are frequently found in eukaryotic proteins involved in basic cell regulatory processes16. The predicted protein also contains one stretch of basic residues (amino acids 20–30) that resembles a nuclear localization signal17 (Fig. 1d). Several independent transgenic lines homozygous with respect to a construct carrying a translational fusion of FVE to green fluorescent protein (GFP) under the control of FVE regulatory sequences showed nuclear GFP signal in most seedling cells (Fig. 2b).

(a) Col and fve-3 plants grown for 4 weeks under long-day (16-h) or 6 weeks under short-day (8-h) photoperiods with (V+) or without (V−) 8 weeks of vernalization at 4 °C. (b) Total number of leaves produced by wild-type and fve mutant plants under different environmental conditions. Plants were grown under long days or short days without vernalization (white columns) or after vernalization for 4 weeks (light gray columns) or 12 weeks (dark gray columns). (c) Effect of fve mutations on FVE and FLC expression in 12-d-old vegetative plants. Hybridization with an 18S ribosomal probe is shown as loading control. The rubisco large subunit (rbcL) stained with Ponceau is shown as protein loading control. (d) Structure of FVE gene (At2g19520) and protein. Exons are shown as boxes and introns as lines. The predicted translation start (ATG) and stop (TAA) codons are indicated. The 5′ and 3′ end untranslated regions are boxed in black. Mutations in the fve alleles are shown in the upper part, capital letters correspond to the wild-type or mutated nucleotides. In the protein schematic, gray boxes represent WD repeats, the narrow black box represents a putative nuclear localization signal and the asterisk indicates a putative retinoblastoma-binding motif.

(a) Complementation of fve mutants. Flowering time under long-day photoperiod of two representative fve-1 and fve-3 transgenic lines, homozygous with respect to a Ler 5.5-kb FVE genomic construct. (b) Nuclear localization of FVE-GFP fusion protein. DAPI was used for nuclear staining. (c) FVE expression in different organs of Col plants. R, roots; RL, rosette leaves; CL, cauline leaves; St, main stems; Fl, flower buds; Fr, developing fruits. (d) FVE transcript expression in Ler and Col grown in different environmental conditions. LD, long day; SD, Short day; V+, plants vernalized for 4 weeks; V−, unvernalized plants. (e) FVE transcript expression in flowering mutants of the autonomous pathway. (f) Flowering phenotype and FVE expression under long-day photoperiod in transgenic plants overexpressing FVE. 35OX1, 35OX2 and 35OX3 are three representative Ler transgenic lines and 35OX4, 35OX5 and 35OX6 are three representative Col transgenic lines, carrying a single homozygous insertion of FVE cDNA under the 35S promoter. 35OX1 and 35OX4 are cosuppressed lines. DOX1, DOX2 and DOX3 are three representative Ler transgenic lines and DOX4, DOX5 and DOX6 are three representative Col transgenic lines homozygous with respect to a single insertion of a 6.5-kb FVE genomic construct. In a and f, light gray bars show Ler genetic background lines and dark gray bars show Col background lines. In d–f, RNA and proteins were extracted from 14-d-old rosettes. Hybridization with an 18S ribosomal probe and the rubisco large subunit (rbcL) stained with Ponceau are shown as RNA and protein loading controls, respectively.
We analyzed the spatial and temporal expression of FVE at the transcript and protein level (Fig. 2). FVE RNA and protein were detected in all analyzed vegetative organs and in reproductive organs at higher levels (Fig. 2c). Transcript and protein levels were correlated, indicating that the amount of FVE protein is mainly determined by transcriptional regulation. We also analyzed FVE expression in response to different photoperiod or vernalization treatments (Fig. 2d) and in FRI-Sf2, ld, fpa, fca and fy plants with mutations of the autonomous pathway (Fig. 2e) but found no significant differences. Therefore, FVE expression is not strongly regulated during plant development, nor is it affected by mutations in other genes of the pathway or by photoperiod or low temperatures.
Two strategies of genetic manipulation of FVE expression confirmed the correlation between protein amount and transcript levels (Fig. 2f). First, homozygous transgenic plants overexpressing the FVE cDNA from a 35S promoter showed several times more FVE transcript and protein. A high proportion of those transgenic plants (7 of 15 lines) showed cosuppression of FVE expression and had phenotypes similar to fve mutants (Fig. 2f). Second, increasing the number of genomic FVE copies in Landsberg erecta (Ler) or Columbia (Col) transgenic plants caused comparable increases in transcript and protein levels, but no cosuppression was observed. These plants had either a wild-type phenotype or a small reduction in their flowering time when grown under either long days (Fig. 2f) or short days (data not shown). This indicates that FVE overexpression is not sufficient to significantly alter flowering time, even under limiting conditions of flowering induction. Therefore, FVE concentration in the cell seems not to be a limiting factor and FVE function might require other protein factors.
FVE (or AtMSI4) is a plant homolog of yeast MSI (multicopy suppressor of IRA1) and mammalian retinoblastoma-associated proteins RbAp46 and RbAp48 (ref. 15). The A. thaliana genome contains a small gene family of five MSI-like genes18,19,20. Database searches showed that MSI genes are present in all sequenced eukaryotic genomes and overall, plant genomes have more MSI genes than genomes from other kingdoms. A comparative phylogenetic analysis of A. thaliana MSI-like proteins with other similar proteins from plants, yeasts and animals arranged A. thaliana MSI proteins in two main clades20 (Supplementary Fig. 2 online). AtMSI1, AtMSI2 and AtMSI3 are more closely related to animal sequences than the other A. thaliana proteins and, together with other plant proteins, cluster in a large heterogeneous clade. FVE (AtMSI4) and AtMSI5 share a high similarity (75% identity) and are grouped in a clade that includes only plant MSI-like proteins. All these proteins are more similar to one another than to other MSI-like proteins of the same plant species. Hence, divergence among plant MSI-like proteins predates the monocot-dicot divergence. Plant-specific proteins might have been recruited along plant evolution to carry out functions more specifically related to plant biology. Moreover, the specific and distinct phenotypes of AtMSI1 antisense transgenic plants20,21 and fve (AtMSI4) null mutants suggest that they are not functionally redundant, nor are they redundant with other A. thaliana MSI-like proteins.
MSI or retinoblastoma-associated proteins are present in protein complexes involved in chromatin assembly and histone modification20,21. In A. thaliana, AtMSI1 is associated with the CAF complex22 and with PcG complexes, such as MEDEA21. Additionally, MSI-like proteins associate with retinoblastoma in animal and plant systems, and ZmRbAp1, the maize FVE ortholog, interacts in vitro with maize retinoblastoma protein ZmRBR1 (ref. 23). As FVE possesses one putative retinoblastoma-binding motif (L-X-C-X-D)24 in the fourth WD repeat (Fig. 1c), we analyzed the interaction between FVE and retinoblastoma. Yeast two-hybrid assays did not show significant interactions, probably owing to intrinsic limitations of the system when dealing with transcriptional repressors. For this reason, we carried out immunoprecipitation assays to determine whether FVE associates with the ZmRBR1. We added recombinant ZmRBR1 to protein crude extracts from Col wild-type plants, null fve-3 mutants and transgenic A. thaliana plants overexpressing FVE. ZmRBR1 was immunoprecipitated from extracts of plants overexpressing FVE but not of fve-3 plants, indicating an interaction between FVE and ZmRBR1 (Fig. 3) and suggesting that FVE could be part of retinoblastoma-containing complexes in A. thaliana. Furthermore, ZmRBR1 was not immunoprecipitated from extracts of wild-type Col plants (data not shown), probably because all the FVE protein in wild-type plants is part of stable protein complexes and not accessible to ZmRBR1.

(a) An immunoprecipitation assay (IP) using protein extracts prepared from seedlings of null mutant fve-3 and of the 35S:FVE line 35OX5, precipitated with antibody to FVE and hybridized in western blots (WB) with antibody to FVE. (b) An immunoprecipitation (IP) assay in which recombinant ZmRBR1 protein was added to the 35S:FVE and fve-3 protein crude extracts and incubated with antibody to FVE. The proteins precipitated in this way were hybridized in western blots (WB) with antibody to ZmRBR1.
In yeast and animal systems, retinoblastoma proteins can function as transcriptional repressors by recruiting HD1/RPD3 histone deacetylases (HDACs). Retinoblastoma-associated proteins similar to FVE are components of such complexes25. Because FVE function is required for FLC repression, we speculated that FVE could repress FLC expression by participating in HDAC complexes modifying the chromatin structure of FLC. To investigate this hypothesis, we analyzed histone acetylation in FLC using chromatin immunoprecipitation (ChIP) assays (Fig. 4). Chromatin of fve-1, fve-3, fca-1, Ler and Col plants was immunoprecipitated using antibodies against acetylated histones H3 or H4. We amplified ten DNA fragments spanning the promoter, first exon and first intron of FLC (Fig. 4a) from the precipitated chromatin. FLC sequences were consistently more abundant in precipitated chromatin from fve mutants than from wild-type plants in three independent experiments (Fig. 4b; DNA from fve-1 and fve-3 mutants was 1.9 ± 0.2 and 1.9 ± 0.3 times more abundant than that from respective wild-type plants). Comparable but smaller enrichment was observed for acetylated histone H4. In contrast, the same sequences had similar abundances in plants with another mutation of the autonomous pathway, fca-1, and in Ler plants (mean relative fca-1/Ler value = 1.17 ± 0.07). Thus, in fve mutants, FLC chromatin is enriched in acetylated histones, indicating that FVE affects histone acetylation at FLC. We conclude that FVE is required for histone deacetylation of the FLC chromatin in A. thaliana.

(a) FLC genomic region analyzed using antibodies to acetylated H3 and H4. The gray box corresponds to the promoter region, white boxes to exons and black box to the first intron. The different FLC fragments analyzed by semiquantitative PCR are represented and numbered below. (b) PCR products after 28 cycles of Ler wild type and fve-1 mutant using DNA purified from chromatin immunoprecipitated with antibodies against acetylated H3 (AcH3). ACTIN2 was amplified as control for DNA quantification.
MSI proteins are biochemically well characterized in several organisms, where they have been involved in the negative regulation of cell cycle and cell differentiation21,25, but their developmental functions are poorly understood. The identification of FVE as a member of a plant-specific clade of retinoblastoma-associated proteins provides the first genetic evidence to our knowledge of a developmental function for this clade of MSI-like proteins. Furthermore, the participation of FVE in the negative regulation of FLC transcription through histone deacetylation characterizes a new genetic mechanism in the regulation of flowering.
While this manuscript was under review, the gene FLD of the autonomous pathway was shown to encode a homolog of a protein found in mammal HDAC complexes26. Similar to fve, fld mutants show hyperacetylation of histones in FLC chromatin. These findings further support the involvement of histone acetylation in the regulation of flowering time in A. thaliana.
Methods
Plant materials, growth conditions and measurement of flowering time.
We used the following A. thaliana strains: the wild-type laboratory accessions Ler and Col; the fve-1 and fve-2 mutated alleles in Ler background, generated by ethyl methane sulfonate mutagenesis6; the fast neutron–induced fve-3 and fve-4 mutated alleles in Col background; the fca-1, fpa-1 and fy-1 flowering mutations of the autonomous pathway in a Ler genetic background6; and the ld-1 mutation and the FRI-Sf2 allele from Saint Feliu accession introgressed in Ler background27. We measured flowering time as the total number of leaves (rosette and cauline leaves) developed by a plant6.
Molecular characterization of FVE.
We obtained genomic constructs, overexpression cDNA constructs and FVE-GFP fusion constructs using standard procedures as detailed in Supplementary Methods online. We transformed A. thaliana plants by the floral dip method. We analyzed FVE mRNA expression by hybridization of northern blots containing 30 μg of total RNA isolated by standard procedures with the full length FVE cDNA. Similarly, we analyzed FLC expression using as probe an EcoRI-SphI 700-bp fragment of the FLC cDNA3. Northern blots were hybridized with an 18S ribosomal probe as loading control.
We raised rabbit polyclonal antibodies against a chemically synthesized FVE peptide corresponding to a sequence of 26 N-terminal amino acids present only in FVE but not in other A. thaliana MSI proteins. This antibody recognized a protein of ∼56 kDa at equivalent levels in wild-type and fve-1 plants but not in fve-3 plants, showing its FVE specificity. We used this antibody to analyze FVE protein expression by hybridizing western blots containing ∼40 μg of total proteins extracted by standard procedures, resolved on 8% acrylamide SDS gels and transferred to Biotrace NT nitrocellulose membranes (Pall, Gelman). We detected proteins with SuperSignal West Pico Chemiluminiscence Substrate kit (Pierce).
Interaction between FVE and retinoblastoma protein.
We carried out immunoprecipitation assays as described28 using A. thaliana protein extracts prepared from 12-d-old to 14-d-old seedlings grown under long-day photoperiod conditions in solid nutrient medium. We prepared extracts by homogenizing seedlings in ice-cold buffer containing 50 mM Tris Cl, 150 mM NaCl, 0.5% Nonidet P-40, 1 mM phenylmethylsulfonyl fluoride and 1× protease inhibitor cocktail. We precleared extracts by spinning for 15 min in a microcentrifuge. Before immunoprecipitation, we incubated 1 mg of extract with 50 μl of protein A-agarose (SIGMA) for 2 h at 4 °C with gentle mixing followed by brief centrifugation. We then added a total of 10 μl of antibody to FVE to the precleared extract and incubated it for 12 h at 4 °C. To study in vitro the putative interaction between recombinant ZmRBR1 and FVE, we added recombinant ZmRBR1 (ref. 29) to precleared protein extract isolated from the transgenic line 35OX5 (overexpressing FVE under the 35S promoter) and fve-3 plants previously incubated with antibody to FVE. We collected protein complexes between ZmRBR1 and AtFVE after adding 50 μl of protein A-agarose and washed them three times for 15 min in 1 ml of ice-cold washing buffer. We analyzed the immunoprecipitated proteins by western blotting using the antibody to FVE or a rabbit antiserum against the maize retinoblastoma protein. The latter was derived against the recombinant ZmRBR1 A/B pocket25.
ChIP assays and PCR.
We carried out ChIP assays as described30 with minor modifications. We collected 10-d-old Ler, fve-1, fca-1, Col and fve-3 plants, at vegetative stage, grown on nutrient solid medium and cross-linked their chromatin proteins to DNA by formaldehyde fixation. We isolated chromatin and sonicated it to produce DNA fragments of 300–1,500 bp. We then immunoprecipitated chromatin using antibodies against acetylated histone H3 recognizing acetylated Lys9 and Lys14 residues (#06-599 from Upstate Biotechnology) or the antibody to acetylated histone H4 recognizing acetylated Lys5, Lys8, Lys12 and Lys16 residues (#06-866 from Upstate Biotechnology). We then reversed the cross-links by incubation at 65 °C for 5 h. We purified DNA with QIAquick spin columns (QIAGEN) according to the manufacturer's instructions and eluted it in 40 μl of 10 mM Tris-HCl, 1 mM EDTA (pH 8.0).
We carried out semiquantitative PCR to amplify the ten different fragments of the FLC flowering gene3,4 (details and primer sequences available on request). All PCR reactions were done in a final volume of 20 μl using 1 μl of eluted DNA and applying 23–30 cycles (optimized depending on the fragment to avoid saturation of the reaction) of 94 °C (30 s), 56 °C (45 s) and 72 °C (20 s). The amount of amplified DNA was first quantified by image analysis of the DNA bands on agarose gel (visible after 26–30 PCR cycles) using MCID Analysis software (Imaging Research). The results of these quantifications were verified by phosphorimager analysis after hybridization of FLC genomic and ACTIN2 probes on Southern blots containing DNA amplified with 23 cycles (not visible on agarose gels). Data provided in the text are from the first quantification. For each fragment in each genotype sample, the amount of immunoprecipitated DNA was estimated as the amount of amplified DNA normalized to the amount of ACTIN2 DNA amplified30. To estimate the relative amount of immunoprecipitated DNA in the mutants versus the corresponding wild type, we determined the normalized amount of immunoprecipitated DNA in the mutant relative to that of the wild type and calculated the average of the relative value of the ten FLC fragments. We carried out, quantified and averaged three independent experiments including the mutant and wild-type genotypes.
GenBank accession numbers.
A. thaliana Ler FVE genomic sequence, AF498101; A. thaliana Ler FVE cDNA sequence, AF498102.
Note: Supplementary information is available on the Nature Genetics website.
References
Simpson, G.G. & Dean, C. Arabidopsis, the rosetta stone of flowering time?. Science 296, 285–289 (2002).
Koornneef, M., Alonso-Blanco, C., Peeters, A.J.M. & Soppe, W. Genetic control of flowering time in Arabidopsis. Annu. Rev. Plant Physiol. Plant Mol. Biol. 49, 345–370 (1998).
Michaels, S.D. & Amasino, R.M. FLOWERING LOCUS C encodes a novel MADS-domain protein that acts as a repressor of flowering. Plant Cell 11, 949–956 (1999).
Sheldon, C.C. et al. The FLF MADS box gene: a repressor of flowering in Arabidopsis regulated by vernalization and methylation. Plant Cell 11, 445–458 (1999).
Johanson, U. et al. Molecular analysis of FRIGIDA, a major determinant of natural variation in Arabidopsis flowering time. Science 290, 344–347 (2000).
Koornneef, M., Hanhart, C.J. & Van der Veen, J.H. A genetic and physiological analysis of late flowering mutants in Arabidopsis thaliana. Mol. Gen. Genet. 229, 57–66 (1991).
Sanda, S.L. & Amasino, R.M. Interaction of FLC and late-flowering mutations in Arabidopsis thaliana. Mol. Gen. Genet. 251, 69–74 (1996).
Michaels, S.D. & Amasino, R.M. Loss of FLOWERING LOCUS C activity eliminates the late-flowering phenotype of FRIGIDA and autonomous-pathway mutations, but not responsiveness to vernalization. Plant Cell 13, 935–941 (2001).
Macknight, R. et al. FCA, a gene controlling flowering time in Arabidopsis, encodes a protein containing RNA-binding domains. Cell 89, 1–20 (1997).
Schomburg, F.M., Patton, D.A., Meinke, D.W. & Amasino, R.M. FPA, a gene involved in floral induction in Arabidopsis, encodes a protein containing RNA-recognition motifs. Plant Cell 13, 1427–1436 (2001).
Simpson, G.G., Dijkwel, P.P., Quesada, V., Henderson, I. & Dean, C. FY is an RNA 3′ end-processing factor that interacts with FCA to control the Arabidopsis floral transition. Cell 113, 777–787 (2003).
Lee, I. et al. Isolation of LUMINIDEPENDENS: A gene involved in the control of flowering time in Arabidopsis. Plant Cell 6, 75–83 (1994).
Martínez-Zapater, J.M. & Somerville, C.R. Effect of light quality and vernalization on late-flowering mutant of Arabidopsis thaliana. Plant Physiol. 92, 770–776 (1990).
Martínez-Zapater, J.M., Jarillo, J.A., Cruz-Alvarez, M., Roldan, M. & Salinas, J. Arabidopsis late-flowering fve mutants are affected in both vegetative and reproductive development. Plant J. 7, 543–551 (1995).
Kenzior, A.L. & Folk, W.R. AtMSI4 and RbAp48 WD-40 repeat proteins bind metal ions. FEBS Lett. 440, 425–429 (1998).
Li, D. & Roberts, R. WD-repeat proteins: structure characteristics, biological function, and their involvement in human diseases. Cell. Mol. Life Sci. 58, 2085–2097 (2001).
Hicks, G.R. & Raikhel, N.V. Protein import into the nucleus: an integrated view. Ann. Rev. Cell Dev. Biol. 11, 155–188 (1995).
Ach, R.A., Taranto, P. & Gruissem, W. A conserved family of WD-40 proteins binds to the retinoblastoma protein in both plants and animals. Plant Cell 9, 1595–1606 (1997).
The Arabidopsis genome initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815 (2000).
Hennig, L., Taranto, P., Walser, M., Schönrock, N. & Gruissem, W. Arabidopsis MSI1 is required for epigenetic maintenance of reproductive development. Development 130, 2555–2565 (2003).
Köhler, C. et al. Arabidopsis MSI1 is a component of the MEA/FIE Polycomb group complex and required for seed development. EMBO J. 22, 4804–4814 (2003).
Kaya, H. et al. FASCIATA genes for chromatin assembly factor-1 in Arabidopsis maintain the cellular organization of apical meristems. Cell 104, 131–142 (2001).
Rossi, V. et al. The maize WD-repeat gene ZmRbAp1 encodes a member of the MSI/RbAp sub-family and is differentially expressed during endosperm development. Mol. Genet. Genomics 265, 576–584 (2001).
Williams, L. & Grafi, G. The retinoblastoma protein - a bridge to heterochromatin. Trends Plant Sci. 5, 239–240 (2000).
Rossi, V. et al. A maize histone deacetylase and retinoblastoma-related protein physically interact and cooperate in repressing gene transcription. Plant Mol. Biol. 51, 401–413 (2003).
He, Y., Michaels, S.D. & Amasino, R.M. Regulation of flowering time by histone acetylation in Arabidopsis. Science 302, 1751–1754 (2003).
Lee, I., Michaels, S.D., Masshardt, A.S. & Amasino, R.M. The late-flowering phenotype of FRIGIDA and mutations in LUMINIDEPENDENS is suppressed in the Landsberg erecta strain of Arabidopsis. Plant J. 6, 903–909 (1994).
Gray, W.M. et al. Identification of an SCF ubiquitin-ligase complex required for auxin response in Arabidopsis thaliana. Genes Dev. 13, 1678–1691 (1999).
Boniotti, M.B. & Gutierrez, C. A cell-cycle-regulated kinase activity phosphorylates plant retinoblastoma protein and contains, in Arabidopsis, a CDKA/cyclin D complex. Plant J. 28, 341–350 (2001).
Johnson, L.M., Cao, X. & Jacobsen, S.E. Interplay between two epigenetic marks: DNA methylation and histone H3 lysine 9 methylation. Curr. Biol. 12, 1360–1367 (2002).
Acknowledgements
We thank M. Koornneef, F. Schomburg, R. Amasino and Y. Komeda for providing the seeds of fve-1, fve-2, fve-3 and fve-4 mutants, respectively; M. Koornneef and W. Soppe for providing a Ler genomic library; S. Michaels and R. Amasino for a FLC cDNA clone; and C. Gutierrez and J. del Pozo for supplying the ZmRBR1 protein and the corresponding antibody. This work has been supported by grants from the European Union and from the Spanish Ministerio de Ciencia y Tecnología. I.A. was supported by predoctoral fellowship from the Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria. C.A.-B. was supported by a contract “Ramón y Cajal” from the Spanish Ministerio de Ciencia y Tecnología.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
About this article
Cite this article
Ausín, I., Alonso-Blanco, C., Jarillo, J. et al. Regulation of flowering time by FVE, a retinoblastoma-associated protein. Nat Genet 36, 162–166 (2004). https://doi.org/10.1038/ng1295
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng1295
This article is cited by
-
Identification of RPD3/HDA1 Family Genes in Sugar Beet and Response to Abiotic Stresses
Sugar Tech (2023)
-
Expression patterns of HDA9, HDA6 and FLD in Chinese cabbage (Brassica rapa L. ssp. pekinensis) under different photoperiods and their protein interactions
Brazilian Journal of Botany (2023)
-
MicroRNA miR394 regulates flowering time in Arabidopsis thaliana
Plant Cell Reports (2022)
-
Genome-Wide Association Study (GWAS) to Identify Salt-Tolerance QTLs Carrying Novel Candidate Genes in Rice During Early Vegetative Stage
Rice (2021)
-
BrcuHAC1 is a histone acetyltransferase that affects bolting development in Chinese flowering cabbage
Journal of Genetics (2021)
