






Advertisement
-
-
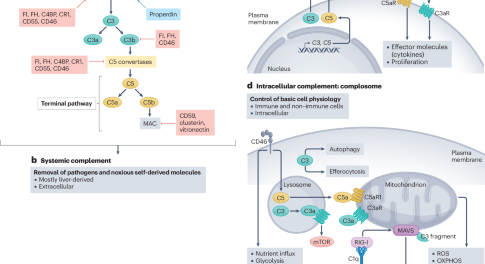
Canonical and non-canonical roles of complement in atherosclerosis
In this Review, Kemper and colleagues discuss the canonical and non-canonical roles of the complement system in the pathogenesis of atherosclerosis and discuss potential new therapeutic strategies targeting the complement system for the prevention and treatment of atherosclerotic cardiovascular disease.
-
-










Anticoagulants
Anticoagulant drugs are used to prevent and treat thrombotic disorders in millions of patients worldwide. This Milestone plots the history of anticoagulant drugs, starting with the discovery and clinical trials of heparin and warfarin.
Advertisement
Trending - Altmetric
-
Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty
-
International Atherosclerosis Society guidance for implementing best practice in the care of familial hypercholesterolaemia
-
Gender medicine: effects of sex and gender on cardiovascular disease manifestation and outcomes
-
Guidelines on ‘added’ sugars are unscientific and unnecessary
Advertisement
