
Abstract
Cell–cell communication and interaction is critical during fertilization and triggers free cytosolic calcium ([Ca2+]cyto) as a key signal for egg activation and a polyspermy block in animal oocytes. Fertilization in flowering plants is more complex, involving interaction of a pollen tube with egg adjoining synergid cells, culminating in release of two sperm cells and their fusion with the egg and central cell, respectively. Here, we report the occurrence and role of [Ca2+]cyto signals during the entire double fertilization process in Arabidopsis. [Ca2+]cyto oscillations are initiated in synergid cells after physical contact with the pollen tube apex. In egg and central cells, a short [Ca2+]cyto transient is associated with pollen tube burst and sperm cell arrival. A second extended [Ca2+]cyto transient solely in the egg cell is correlated with successful fertilization. Thus, each female cell type involved in double fertilization displays a characteristic [Ca2+]cyto signature differing by timing and behaviour from [Ca2+]cyto waves reported in mammals.
Similar content being viewed by others


Introduction
Fertilization in eukaryotes involves species-specific recognition, local interaction, attachment and eventually fusion of two gametic cells of opposite sex. The fertilization process in flowering plants (angiosperms) is especially complex and requires communication between the cells of two haploid gametophytes1. The pollen tube comprises the male gametophyte and consists of a vegetative tube cell that carries two immotile sperm cells. The female gametophyte is composed of the two female gametes (egg and central cell) and accessory cells at opposite poles. Two synergid cells adjoining the egg cell are located at the micropylar entry of the ovule, while antipodal cells neighbouring the central cell are found at the chalazal pole. After sperm cell delivery and fusion of two gamete pairs, a process termed as double fertilization, the fertilized central cell develops into the endosperm and the fertilized egg cell gives rise to the embryo. Numerous cell–cell communication events take place during the attraction of the male towards the female gametophyte and their mutual interaction. It was reported that synergid cells are responsible for pollen tube attraction by secreting species-specific chemotactic peptides2,3,4,5. Moreover, they recognize arriving pollen tubes involving cell surface receptors6,7,8 and control sperm release9,10. After pollen tube burst, the two sperm cells rapidly reach the cleft between egg and central cell representing the site of gamete interaction and fusion11. Sperm cells interact with female gametes and become activated by egg cell-secreted small proteins12. The molecular mechanisms by which the male and female gametes recognize each other and the intracellular signalling events that result in synergid cell death, female gamete activation, control of successful plasmogamy and the prevention of multiple sperm fusion (polyspermy) remain to be identified.
In animals, calcium plays a key role during fertilization as a secondary messenger of cell–cell interaction events and triggers, for example, the establishment of a fertilization membrane to prevent polyspermy13,14,15. While the role of calcium signalling during double fertilization in flowering plants still remains to be unveiled, it is known that calcium signalling plays a key role in cell–cell recognition, for example, during the innate immune response to pathogens or the establishment of symbiosis16,17. Calcium has also known functions during the early stages of fertilization, which include promoting pollen tube growth, guidance and eventually the regulation of its burst18,19,20,21. Recently, it was also reported that the arrival of the pollen tube causes [Ca2+]cyto oscillations in synergid cells20. This observation opened up the question how the pollen tube and the synergid cells interact with each other to trigger these oscillations, whether this occurred through direct contact or through signals that act at a distance.
The first evidence that calcium indeed is also involved during gamete fusion was obtained from in vitro fertilization experiments. Gamete fertilization in maize mediated by calcium chloride was shown to occur in a cell-specific manner and leads to a block of polyspermy22. More detailed studies have reported that Ca2+ influx occurred in this in vitro system after egg–sperm fusion and spreads as a wave front from the fusion site towards the whole cell lasting for up to 30 min (refs 23, 24, 25). The authors further observed that fusion, but not gamete adhesion triggered a transient [Ca2+]cyto rise in fertilized egg cells and found that cell wall material is formed after fusion likely representing a block to polyspermy functionally similar to the fertilization membrane in animals. These data strongly support the notion that calcium signalling may be of similar importance during double fertilization in plants compared with fertilization in animals. However, in vitro investigations are limited as double fertilization mechanisms involve many timely and accurately regulated cellular interactions to guarantee reproductive success.
To visualize when and how calcium transients are triggered during the whole double fertilization process in vivo, and to enable studies on the functional role(s) of [Ca2+]cyto changes during fertilization, we used an enhanced genetically encoded fluorescent calcium sensor. This sensor consists of a calcium-binding domain fused to two fluorophores that are spectrally overlapping. Their proximity and orientation depends on the conformation of the binding domain. Calcium binding therefore results in differential Förster Resonance Energy Transfer (FRET) between the donor and acceptor fluorophores. Most widely used in plants are Yellow Cameleon sensors26,27,28,29 that contain a calmodulin-based calcium-binding domain. It has been argued, however, that due to the high degree of conservation of calmodulin, cellular pathways might interfere with the sensor, and vice versa30. This led to the development of the calcium sensor CerTN-L15, a sensor with enhanced folding, increased brightness and a calcium-binding domain based on troponin C, a protein exclusively found in myocytes31. Thus, it is unlikely that CerTN-L15 exhibits strong interactions with plant proteins.
By using a modified semi-in vivo system32 and expressing the improved calcium sensor CerTN-L15 from various female gametophyte cell-type-specific promoters, we were able to monitor [Ca2+]cyto signatures by live-cell imaging throughout the whole double fertilization process in the model plant Arabidopsis. We report that direct contact with the pollen tube apex and not its shank region triggers polarized [Ca2+]cyto oscillations in synergids cells. Furthermore, we link sperm cell dynamics at the gamete fusion site to specific Ca2+ signatures in both egg and central cells. We show that first Ca2+ transients precede fertilization, indicating that they might be required for early events such as female gamete activation. A second Ca2+ transient solely in the egg cell appears associated to sperm–egg fusion and might trigger later events such as a block to polyspermy.
Results
Sensor based recording of cell-specific calcium changes
To monitor [Ca2+]cyto during the whole double fertilization process, we expressed the Ca2+ sensor CerTN-L15 (ref. 31) under control of cell-type-specific promoters in synergid cells (MYB98-promoter; AT4G18770 (ref. 33)), egg cell (EC1.1 promoter; AT1G76750 (ref. 12)) and the central cell (DD65-promoter; AT3G10890 (ref. 34)) as indicated (Fig. 1a). This sensor is based on the Ca2+−binding domain of troponin C and two spectrally overlapping fluorescent molecules cerulean and citrine that act as donor and acceptor for FRET, respectively. As the CerTN-L15 sensor has not been used in plants before, we first validated its functionality in roots expressing the sensor constitutively under control of a 35S promoter, using a microfluidic device that allows imaging under controlled perfusion of roots35. Repeated pulsed treatments with a hyperosmotic solution resulted in [Ca2+]cyto signatures with a minor increase in FRET ratio during the treatment-induced root shrinkage and a steep incline of the FRET ratio after wash-out and return to original root length, followed by a gradual return of the FRET ratio to the baseline (Supplementary Fig. 1). This characteristic signature has been observed before using Yellow Cameleon sensors29,36 indicating that CerTN-L15 is functional in plants.

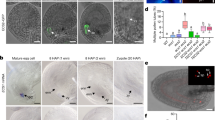
(a) Expression of the calcium sensor CerTN-L15 in specific cells of the female gametophyte. Left panel: schematic of an ovule and approaching pollen tube (PT, orange) containing two sperm cells (SP, red) and the vegetative tube nucleus (indicated in blue). The female gametophyte consists of two synergid cells (SY, yellow), one egg cell (EC, green) and one central cell (CC, cyan). Right panels: merged bright field and confocal images of ovules expressing CerTN-L15 in specific cell types (promoters are in parentheses): SY (pMYB98), EC (pEC1) and CC (pDD65). (b) Arrival and discharge of a PT at the SYs. Time series of a PT of the LHR marker line displaying the tube apex (outline of the PT is indicated by dotted lines) approaching SYs (false colour ratiometric images of pMYB98:CerTN-L15). SPs (arrowheads) of this marker line are discharged about 62 min after recording was initiated. The following stages are visible: arrival of a PT (6.8 min), first PT contact with SY (13.5 min), before (24.2 min) and during (24.5 min) an oscillation of [Ca2+]cyto, growth of PT around SY (45.0 min, 55.0 min), PT shortly before (61.8 min) and after (63.3 min) release of SPs. See corresponding Supplementary Movie 1. (c) [Ca2+]cyto signature of SYs. Normalized ratio changes of CerTN-L15 in cells shown in b. Arrowheads indicate time points of images in b. Red arrowhead: first physical contact of PT and SY, diamond: pollen tube burst. (d) Variations of [Ca2+]cyto signatures in SYs. Selection of representative [Ca2+]cyto measurements in SYs in the presence (+PT, upper panel) or absence of a PT (−PT, lower panel). Note that PT burst occured only in upper example showing high [Ca2]cyto amplitudes. Red arrowhead marks the time point where the first physical contact of a PT and the synergid could be observed, diamond indicates pollen tube burst. The first graph of the lower panel (−PT) shows typical [Ca2+]cyto recording of SY without PT. The second graph displays an exceptional case of spontaneous [Ca2+]cyto oscillations in SY without a PT being present. This bahaviour was observed in two out of 13 cases. Scale bars, 20 μm.
Pollen tube apex triggers calcium signatures in synergids
To record [Ca2+]cyto signatures in cells of the female gametophyte, we performed our measurements on excised Arabidopsis ovules arranged around a pollinated pistil32. This semi-in vivo setup was modified to achieve automated time-lapse imaging at high spatiotemporal resolution. When CerTN-L15 was expressed in synergid cells, we observed repeated FRET ratio changes consistent with [Ca2+]cyto increases in 23 out of 25 experiments when the pollen tube successfully interacted with the synergid cells (Fig. 1b,c). The [Ca2+]cyto transients occurred with variable periodicity within and among cells with a time interval of 100–200 s and durations of 50–170 s per individual transient. (Fig. 1d, Supplementary Fig. 2a, Movies 1 and 2). Burst of pollen tubes and receptive synergid cell occurred between 30 and 50 min after interaction with synergid cells. [Ca2+]cyto oscillations with low amplitude (lower graphs in Fig. 1d) did not result in burst. When pollen tubes failed to target the micropyle of the ovule or did not reach the synergid cells, we detected spontaneous weak [Ca2+]cyto fluctuations in two out of 16 trials (Fig. 1d, lower panel). In both cases, fluctuations were less regular and showed significantly reduced amplitude by comparison with the signatures observed when pollen tubes successfully approached the synergid cells. We conclude from these observations that limited [Ca2+]cyto oscillations can occur spontaneously in synergid cells at a low likelihood. Only pollen tubes that successfully enter the ovule and interact with the synergid cells induce high persistent and effective [Ca2+]cyto oscillations.
Consistent with recently published work37, we observed that the pollen tube did not burst in the filiform apparatus as suggested previously (reviewed in ref. 37). Instead the pollen tube grew for up to 60 min in close proximity and around the synergid cells towards the gamete fusion site before rupture and synergid cell death occurred (Supplementary Movie 1).
To investigate whether communication between the pollen tube and the synergid cells occurs already at a distance, or if physical cell–cell contact is required, we monitored the onset of Ca2+ oscillations as a function of the distance between the two cells. To this end, we visualized the pollen tube apex by developing a novel marker (PLAT52:RemCA-tagRFP) targeting tagRFP with the carboxyl-terminal (C-terminal) anchor sequence of remorin to the plasma membrane of pollen tubes and introduced it into a homozygous sperm nuclei marker line (PH3.3:H3.3-mRFP38) resulting in the double marker line LHR (Lat52:tagRFP-T-REM; HTR10:HTR10-mRFP; see Methods for details). In all eight measurements where the onset of [Ca2+]cyto oscillations in synergid cells and the advance of the pollen tube tip could be monitored simultaneously, the first substantial [Ca2+]cyto transients (ratio change >5σ of baseline of first derivative, see Methods) were detected when the distance between the two cells could no longer be resolved (Fig. 1b,c). In three of the 25 experiments, the position of the two synergid cells allowed separate measurements of cytoplasmic CerTN-L15 fluorescence within each cell. We observed that the amplitude and frequency of [Ca2+]cyto oscillations depended on the relative distance and position of the pollen tube tip and either of the two synergid cells. Only the synergid cell in direct contact with the pollen tube tip showed continuous [Ca2+]cyto oscillations (Fig. 2a,b).

(a) CerTN-L15 signals observed in each of the two SYs separately. Left panel: merged confocal image of a LHR PT tip (red) in contact with both pMYB98:CerTN-L15 expressing SYs (YFP signal of CerTN-L15 in green). Both SYs can be seen separately (SY1 and SY2). Right panels: one representative [Ca2+]cyto transient (beginning, maximum, end) occurring only in SY1, which is in contact with the PT. LHR pollen is shown in grey scale, ratio changes of CerTN-L15 in SYs are displayed as heat map. (b) Ratio changes recorded for both SYs. The ratio changes of CerTN-L15 were measured in the two SYs separately. Arrowhead marks the [Ca2+]cyto transient shown in a. (c) Local [Ca2+]cyto transients in relation to PT tip position. Two [Ca2+]cyto transients of one time series are shown. [Ca2+]cyto transients with LHR PT tip in contact with the filiform apparatus (upper row) and after being grown around the SYs (lower row). Left panel in both rows: merged confocal image of a LHR PT tip (red) in contact with pMYB98:CerTN-L15 SYs (YFP signal of CerTN-L15 in yellow). Right panels of both rows: time points of corresponding time series before, during and after a transient of [Ca2+]cyto in the SYs (ratio of CerTN-L15 displayed in pseudo colour). The maximum of the [Ca2+]cyto is different in both rows and correlates with the position of the PT tip (dotted line indicates the position of the PT according to the signal shown in the left panel). (d) Mapping of amplitudes of [Ca2+]cyto transients in relation to the distance to PT tip. Schematic of a PT tip in contact with a SY. Amplitudes of [Ca2+]cyto elevations were measured in 44 mm regions (indicated as squares) over the SY in time series registered to the PT tip. Measured amplitudes of the maximum number of detected [Ca2+]cyto transients in five independent experiments. Numbers in each square display the normalized average amplitude height. For statistical data, see Supplementary Fig. 2b. Scale bars, 20 μm.
To support this finding and to investigate whether the synergid [Ca2+]cyto signatures represent global or localized responses, we analysed 78 [Ca2+]cyto peaks and detected the highest amplitude in immediate proximity to the contact site of synergid cell and pollen tube tip (Fig. 2c). Progressing pollen tube growth resulted in relocation of the local [Ca2+]cyto maximum that matched the shifted site of contact with the pollen tube tip. To measure whether indeed only the apex or the entire surface of the pollen tube has the capability to elicit a Ca2+ signature in the synergid cells, we mapped the local amplitudes [Ca2+]cyto peaks (n=78) in synergid cells in 4 × 4 μm bins across the whole cell. We found that the sensor amplitude gradually decreased with increasing distance from the pollen tube tip (Fig. 2d, Supplementary Fig. 2b), consistent with the observation that [Ca2+]cyto is highest at the site where the cells are in direct contact. Moreover, we found that the local sensor amplitude in the synergid cell was the highest in closest proximity to the pollen tube apex and not the shank of the tube (Fig. 2d, Supplementary Fig. 2b).
Taken together, the separate measurements of neighbouring synergids and the correlation of pollen tube position and localized synergid response demonstrate that physical contact specifically with the pollen tube apex is necessary to induce and maintain [Ca2+]cyto oscillations in the synergid cell. Our measurements in synergid cells further show that there exists not just one excitable zone, but rather large areas of the synergid cell surface are capable of sensing contact with the pollen tube.
Sperm cell delivery causes calcium signals in female gametes
Once the pollen tube has ruptured and released its content including the two sperm cells, the process of fertilization enters the next stage, the specific interaction and fusion of the two sperm cells with the egg and the central cell, respectively. To investigate whether the interactions between male and female gametes also invoke Ca2+transients and whether there is a correlation between calcium signatures and sperm cell movement, we measured [Ca2+]cyto in egg and central cells, separately.
When measuring CerTN-L15 signals in the central cell during successful pollen tube rupture (n=14), we observed a sharp increase in [Ca2+]cyto lasting for approximately 1 min immediately following pollen tube burst and deformation of the central cell in the area of sperm arrival due to explosive release of pollen tube contents (Fig. 3a,b, t=0 min; Supplementary Movie 3). Beyond this short transient, significant [Ca2+]cyto transients were not detectable, independent of fertilization success (see further examples in Supplementary Fig. 3).

(a) Time series showing arrival of labelled SPs (indicated by arrow heads; LHR marker line) at the gamete fusion site and subsequent plasmogamy with the central cell expressing pDD65:CerTN-L15 (yellow). Images show major steps of the gamete fusion process: directly before PT rupture (0.0 min), after PT rupture and SP arrival (0.3 min), during arrest phase of SPs at the fusion site (5.0 min) and after plasmogamy showing the movement of SP nucleus towards CC nucleus (21.5 min). Scale bar, 20 μm. See also Supplementary Movie 3. (b) [Ca2+]cyto transients in the CC during fertilization. Normalized ratio of CerTN-L15 in CC of time series shown in a. Arrowheads indicate time points corresponding to images shown in a. A single ratio change of 27% occurs during SP release (time point 0.3 min). (c) Time series of a LHR pollen fertilizing an ovule expressing pEC1:CerTN-L15 in the egg cell (EC, yellow). From −17.0 to 0.0 min) the PT is in contact with SYs (not visible). SPs (red, marked by arrowheads) release occurred at 0.0 min. Arrest of SPs is visible at the fusion site (4.0 min), movement of one SP nuclei inside egg cytoplasm (6.8–11.5 min) is followed by the separation of SP nuclei (11.5–35 min) indicating fertilization success. Scale bar, 20 μm. See corresponding Supplementary Movie 4. (d) [Ca2+]cyto signals in the EC during fertilization. Normalized ratio changes of CerTN-L15 in the EC of time series shown in c. Arrowheads with time correspond to the images shown in c. Ratio changes can be seen directly after SP release (0.0 min) and during the arrest phase of the SPs at the fusion site (6.8 min) but not after SP separation (11.5–35.0 min).
In the egg cell, we also observed an immediate deformation of its shape that coincided with the pollen tube burst and the rapid delivery of sperm cells (Fig. 3c). Analysis of [Ca2+]cyto levels during these events (n=26) revealed again a sharp increase that coincided with cell deformation and sperm cell arrival (Fig. 3d, t=0 min). In some experiments, deformation of the egg cell was caused by the growing pollen tube (Fig. 3c, arrows at time points −17 min and −8 min). However, in these cases, calcium transients were never detected (Fig. 3d; Supplementary Figs 4a,b and 6b, Supplementary Movies 4, 5). This indicates that mechanical stimulation of the egg cell is not sufficient to cause the detected elevation of [Ca2+]cyto..
While the sharp [Ca2+]cyto peak at t=0 occurred in both female gametes simultaneously during pollen tube burst, an additional extended [Ca2+]cyto transient with more gradual rise and fall times was detected only in the egg cells (Figs 3d and 4c, Supplementary Figs 3 and 6c). This second [Ca2+]cyto transient appeared on average 3.8 min (±1.9, n=12) after the pollen tube burst and lasted on average for 3.1 min (±0.66, n=12). The second [Ca2+]cyto transient was detected in all successful fertilization events (n=10) as shown in Fig. 4c. When fertilization was not successful, the second [Ca2+]cyto transient could not be detected in 13 out of 15 measurements (Fig. 4d, Supplementary Fig. 4a,b, Supplementary Movie 5). This indicates a functional link between the second extended [Ca2+]cyto transient and successful fertilization.

(a) Left: scheme depicting SP release from a PT growing around receptive SY (drawn in grey). After burst, SPs are located at the fusion site between EC and CC. Upper panel: time series of LHR SP (red, arrowheads) fertilizing an egg cell expressing the CerTN-L15 sensor (yellow). In the left micrograph, zero and orientation of the coordinate system in b are indicated. Lower panel: scheme of different phases of sperm cell movement before and during fusion. Phase I: parallel movement of sperm cells after arrival along fusion site. Phase II: sideward shift of SPEC (single asterisk) towards EC. Phase III: arrest of SPCC and movement of SPEC nucleus towards EC nucleus. Phase IV: sideward shift of SPCC (double asterisk) and subsequent fast sperm nuclei separation. (b) Tracking movement of both sperm cells at fusion site of experiment shown in a. Different colours of the tracks indicate the four different phases and numbers indicate end points of each phase after sperm delivery in minutes. Shift of SPEC occurs at 1:40 min (single asterisk) and shift of SPCC at 14:20 min (double asterisk) in this example. (c) [Ca2+]cyto signals in the EC during successful fertilization. Graphs are normalized and aligned to time of PT rupture (0.0 min). Asterisks mark shift of SPEC, arrowheads mark fast separation of both sperm nuclei indicating successful fertilization of both female gametes. In traces 4 and 5, SPs were not visible at all times. In all cases, two Ca2+ transients occur: a short single transient during PT rupture and a longer extended transient after SPEC shift (Phase II). (d) Variety of [Ca2+]cyto signals in the EC during unsuccessful fertilization. Only a single Ca2+ transient occurs on pollen tube rupture (0.0 min). (e) Exceptional case of EC fertilization. Ratio change of pEC1:CerTN-L15 is shown. SPEC displays two shifts (asterisk) before fusing with the EC. Each shift is followed by a Ca2+ transient in the EC (Corresponding kymograph see Supplementary Fig. 4d). (f) Correlation of SPEC shift towards EC and beginning of second Ca2+ transient in EC (n=10). Formula of linear regression and calculated coefficient of determination (R2) are given. (g) Diffusion of sensor molecules into sperm cells upon gamete fusion. Time points before (top row) and after SPEC shift (bottom row). Left column, fluorescent micrographs of egg cell (yellow, citrine signal of CerTN-L15) and LHR sperm cell nuclei (red). Middle column, false colour representation of egg cell citrine signal. The dotted line displays the egg cell outline before the SPEC shift. The arrowhead indicates spreading of citrine signal towards the position of sperm cells. Scale bars, 20 μm.
Egg cell-specific calcium signature during plasmogamy
To reveal the role of the second [Ca2+]cyto transient for plasmogamy, we analysed the timing of associated events in more detail. In animal oocytes, a [Ca2+]cyto elevation occurs after the sperm–egg fusion. In plants, two female gametes are fertilized by two sperm cells. This double fertilization event is supposed to require molecular events such as cell–cell recognition, gamete attachment, sperm cell separation, gamete membrane fusion, egg activation and cell wall modifications that take place between both male–female gamete pairs. Any of these processes may involve calcium signalling. While it is challenging to directly address each process separately, the timing of events before fertilization can be dissected by using a marker line that labels sperm nuclei and tracking their movement at the fusion site11. Since both sperm cells do not possess any propelling mechanism, their movement presumably results from elastic deformations and pressure exerted by neighbouring cells or differential surface interactions. Hamamura et al.11 reported that sperm delivery is composed of three steps. They are delivered as a pair (i) and after a period of immobility lasting about 7.4 min (ii) they both fuse almost simultaneously about 8.5 min after release with either female gamete without any preference (iii).
To correlate the second [Ca2+]cyto transient with stages of sperm delivery and fusion, we tracked the sperm nuclei after pollen tube rupture and analysed their trajectories for consistent movement patterns. The sperm cell, which eventually fused with the egg cell, was termed SPEC, while the sperm cell that would fuse with the central cell was termed SPCC (Fig. 4a). After pollen tube rupture, we observed four distinct phases of sperm movement, which typically occurred over a travelled distance of 10 μm (Fig. 4b): after delivery, both cells moved initially quickly together along parallel trajectories (phase I, Fig. 4b, orange trajectories); a second phase is initiated by a sudden sideward movement of SPEC apart from SPCC (Fig. 4a,b, single asterisks) followed by slow movement, rotation and temporary arrest (phase II, Fig. 4b, green), while SPCC movement was also slowed down (phase III, Fig. 4b, blue); after a second sudden positional shift, now of SPCC (Fig. 4a,b, double asterisks), both separated sperm nuclei then made final movements inside the female gametes (phase IV, Fig. 4b, violet) indicating fertilization success. Plotting the intensities of each SP in an overlaying kymograph visualizes their velocity over time (Supplementary Fig. 4c). After the arrival of both cells (t=0), an initial rapid parallel movement (orange phase), followed by a slow down and the eventual separation due to backwards movement of SPCC is visualized. The occurrence of both shifts of SPEC and SPCC in opposite directions is clearly visible in all successful fertilization attempts (n=10) but absent when fertilization failed (n=13).
The shifts were also observed in both failed fertilization attempts where the second broader [Ca2+]cyto transient was detected but the sperm nucleus did not continue to move towards the nucleus of the female gamete (data not shown). In a single measurement, where delayed or incomplete plasmogamy was observed, we could detect two broader [Ca2+]cyto transients in the egg cell and also two sudden shifts of SPEC (Fig. 4e, Supplementary Fig. 4d). These exceptions indicate that the shift can occur repeatedly and therefore could precede successful gamete fusion, eventually representing the event of initial sperm cell adhesion to the female gamete before fusion occurs.
To correlate the [Ca2+]cyto signatures in egg cells and the sperm cell movement patterns, we further analysed the occurrence and timing in all measurements where both events could be unequivocally detected (n=10). We found that the sudden movement of SPEC at the end of sperm movement phase (i) generally coincided with the onset of the second broader [Ca2+]cyto transient (R2= 0.9894; time difference between shift and onset of [Ca2+]cyto transient was 5±13 s; Fig. 4f, Supplementary Movie 4). We interpret this finding as strong evidence for a functional link between gamete interaction, Ca2+ signalling in the egg cell and successful plasmogamy.
To determine the time point of plasmogamy as precise as possible, we therefore followed the rational that upon fusion of the plasma membranes of both gametes, cytosolic calcium sensor molecules would immediately diffuse into the sperm cell cytoplasm resulting in detectable citrine fluorescence around the sperm cell nucleus. In four measurements of successful fertilization, measurements were possible as egg cell and SPEC were positioned in the focus plane such that both the fluorescent signals overlapped only minimally. In all four experiments, we were able to detect the spreading of citrine fluorescence signal from the egg cell towards the direction of the sperm cell position immedeately after the SPEC shift, coinciding with the broader [Ca2+]cyto transient (Fig. 4g, Supplementary Fig. 5). In summary, this observation indicates that all three processes, sperm cell shift, onset of the broad [Ca2+]cyto transient and gamete fusion occur all in concert and within less than 20 s.
Discussion
Innovations in specimen handling and application of a sensitive calcium reporter have allowed live-cell observations and detection of cell-type-specific Ca2+ signatures during the entire double fertilization process in the model plant Arabidopsis. Measuring [Ca2+]cyto in vivo in the female gametophyte at high resolution was previously hampered due to the low accessibility of the deeply embedded cells for imaging. We have optimized a previously reported semi-in vivo fertilization assay that allows live-cell observations by using excised ovules and semi-in vitro grown pollen tubes32. Adaptation of this system for confocal fluorescence imaging and expression of sensors driven by cell-specific promoters resulted in reduced background and improved sensitivity. We decided to use the troponin C-based sensor CerTN-L15 as it is expected to be less affected by or interfering with cellular processes as it is the case for calmodulin-based sensors31. The improved brightness and high sensitivity of this sensor also present significant advantages for imaging through several cell layers31,39,40 and thus enabled us to record changes in [Ca2+]cyto inside the ovule at subcellular resolution.
The described experimental setup allowed studies yielding strong evidence for a functional role of Ca2+ signalling during virtually all steps of the double fertilization process in angiosperms. We were able to confirm recently reported [Ca2+]cyto oscillations in synergid cells during pollen tube interaction20 and provide further insights into cell–cell communication between these cells and the provoked [Ca2+]cyto transients. One open question was, for example, whether the communication between the pollen tube tip and synergid cells occurs already at a distance as concluded earlier20 or whether direct cell–cell contact is required. Our measurements show that irregular [Ca2+]cyto elevations can occur spontaneously in synergid cells at a low likelihood, but do not cause cell death. In addition, we observed that only pollen tubes that successfully enter the ovule and interact with the synergid cells induce high persistent [Ca2+]cyto oscillations that may ultimately trigger synergid cell death and pollen tube burst. Usage of a novel marker for the pollen plasma membrane further indicated that physical contact of the pollen tube apex was necessary to induce and maintain [Ca2+]cyto oscillations only in the synergid cell, which was in direct contact with the tube apex. Recently components of the FERONIA (FER) signalling pathway were shown to be involved in the initiation of calcium oscillations and for coupling these with programmed cell death41.
Stimulus-induced [Ca2+]cyto oscillations have been frequently observed in plants. It was reported that their period, amplitude and waveform encode information about stimulus and trigger required to activate specific signalling responses, to regulate protein activity and eventually to alter gene expression42. The spiking nature of the synergid transients is typical for rapidly released Ca2+ from intra- or extracellular stores followed by slower removal43. Observations of comparable [Ca2+]cyto spiking signatures in legume root hairs, for example, induced by symbiotic infection with rhizobial bacteria or mycorrhizal fungi, identified the origin of the [Ca2+]cyto wave at the cell nucleus44,45,46 and therefore depend on specific metabotropic receptors at the plasma membrane that perceive and convey the extracellular signal47. In contrast, during physical stimulation of root epidermal cells, observed [Ca2+]cyto transients originated from the site of stimulation at the cell periphery48, suggesting extracellular stores or peripheral ER as sources of Ca2+ and possibly the involvement of stretch-activated calcium channels49. The localized transients of [Ca2+]cyto in synergid cells also occur in immediate proximity to the cell contact site, which is reminiscent of mechano-sensing. The oscillation signature, however, is similar to that observed during symbiotic infections and thus suggests an involvement of metabotropic receptors and the existence of feedback mechanisms of Ca2+-induced Ca2+ release50. The regulation of such oscillatory increases of [Ca2+]cyto commonly occurs through the receptor-mediated activation of phospholipase C, which increases the levels of inositol-1,4,5-trisphosphate (InsP3) that further triggers InsP3-receptor-mediated Ca2+ release from internal storages.
The signalling cascades that might be provoked by [Ca2+]cyto increase in the synergid cells are currently unknown. During self-incompatibility in poppy, it was shown that [Ca2+]cyto increase is correlated with the generation of reactive oxygen species and NO signalling resulting in cell death. Downstream targets include, for example, components of the cytoskeleton and mitogen-activated protein kinases51. Both synergid cells undergo cell death either during pollen perception or after successful fertilization52,53. We observed that only synergid cells showing high and continuous amplitudes burst during pollen tube interaction. This finding supports previous reports also in animal cells showing that high sustained [Ca2+]cyto levels are toxic to cells and may result in cell death both during necrosis and apoptosis54. The identification and analysis of calcium-dependent signalling components in synergid cells will now be an exciting area of future research activities. Reactive oxygen species were recently reported to mediate pollen tube burst in [Ca2+]cyto-dependent processes55 suggesting the existence of similar mechanisms during synergid cell death. It will be of similar importance to identify the pollen tube apex-derived stimulus and synergid cell surface receptors involved in triggering the described [Ca2+]cyto oscillations. Our results on [Ca2+]cyto oscillations support the notion of a short-range signal secreted from the pollen tube apex that is perceived by receptor(s) of synergid cells triggering a feedback mechanism that cause localized, oscillating release of Ca2+. The FERONIA receptor-like kinase and the glycosylphosphatidylinositol-anchored LORELEI protein are candidate synergid cell surface proteins that have previously been shown to be required for pollen tube burst6,7. Moreover, a secreted peptide, RALF (rapid alkalinization factor) was recently shown to activate FERONIA in the root56. Whether RALF peptides secreted from the pollen tube apex activate FERONIA at the surface of synergid cells inducing [Ca2+]cyto oscillations remains to be determined. Further studies should also involve mutants with defects in phospholipase C/InsP3 signalling and metabotropic receptors (see above). Altogether, these mutants will be instrumental in elucidating the functional importance of the localized [Ca2+]cyto oscillations in the synergid–pollen tube interaction.
During the next act of plant double fertilization, the fusion of sperm cells with both female gametes, an involvement of Ca2+ transients was long suggested by pioneering work applying in vitro fertilization studies with maize egg and sperm cells23,24,25. It was reported that Ca2+ influx occurred after egg–sperm fusion and spreads as a wave front from the fusion site towards the whole cell. However, these studies did not allow to image [Ca2+]cyto dynamics during the whole fertilization process and thus overlooked, for example, the first [Ca2+]cyto transient in both female gametes that is correlated with sperm cell release. The function of this first elevation remains unclear. It might also be correlated with mechanic deformation of female gametes, albeit we did not observe [Ca2+]cyto transients in egg cells, which were occasionally deformed by pollen tubes (see above). It was, however, recently shown that egg cells secrete EC1 protein after pollen tube burst to activate sperm cells and thus enable them to fertilize female gametes12. Thus, the egg cell itself requires activation shortly after the sperm cell release. Further studies will elucidate whether egg activation at this phase requires Ca2+ signalling. Mammalian egg cells are activated by a series of [Ca2+]cyto oscillations that occur shortly after egg–sperm membrane fusion (for review, see ref. 57), thus differing significantly from the single elevation that we have observed during pollen tube burst. A major difference between previously reported in vitro and our semi-in vivo studies is the length of [Ca2+]cyto elevations. It was reported that the [Ca2+]cyto transient after in vitro gamete fusion lasted for up to 30 min, while we observed that the comparable second extended [Ca2+]cyto transient lasted on average only for 3 min. Moreover, the signature of this [Ca2+]cyto transient is different and seems to consists of a few very short spikes, which could not be resolved individually. In agreement with the above-mentioned in vitro studies, we conclude that this [Ca2+]cyto transient was provoked by fusion rather than adhesion. A functional role of the reported Ca2+ influx could not be addressed during in vitro studies. We were able to ultimately correlate the second [Ca2+]cyto transient with plasmogamy and fertilization success. In a parallel study, Hamamura et al.58 found highly similar [Ca2+]cyto signatures during egg cell fertilization using the calcium sensor YC3.60. In line with our findings, the authors also identified a correlation between the occurrence of the second [Ca2+]cyto transient and successful fertilization. In a mutant of the HAP2/GCS1 gene, which has known roles in pollen tube guidance and fertilization, sperm cells were delivered properly but gamete fusion never occurred. Consistently, the authors found that in this mutant the second [Ca2+]cyto transient in the egg cell was not detectable. It will, however, remain challenging to elucidate the exact role of Ca2+–dependent signalling during this short fertilization phase.
In mammals, two separate fusion phases are distinguished after gamete adhesion takes place: a marginal and a separating phase59. During the marginal phase, fusion is initiated and the second phase, which has a different topological character, fusion is completed during a process similar to endocytosis. We suggest that this process is similar in plants. In accordance with the topology of the marginal phase, we observed egg cell cytoplasm extending towards the sperm cell followed by a quick shift of the sperm cell towards the inside of the egg, which might be consistent with the separating phase characterized by endocytosis at the plasma membrane. Moreover, we have observed few cases that showed additional [Ca2+]cyto elevations displaying the signature of the extended second transient. We assume that fusion was initiated in these cases, but not completed allowing additional fusion events associated with [Ca2+]cyto transients. Markers labelling the plasma membrane and cytoskeleton of gametes in combination with Ca2+ sensors whose signals can be optically separated from the gamete surface will help to explain these observations. While diffraction-limited confocal imaging of fertilization presents a significant advance, unequivocal visualization of membrane fusion may also require substantially improved spatial resolution. It also remains to be determined which downstream responses are triggered by these relatively short [Ca2+]cyto transients. It is very likely that the structure of the cytoskeleton undergoes local changes and reorganization to initiate the first fusion phase and to enable the transport of the sperm nucleus towards the egg nucleus. It was also discussed that [Ca2+]cyto transients initiate the formation of cell wall material after fusion likely representing a block to polyspermy functionally similar to the fertilization membrane in animals24. In animal oocytes, [Ca2+]cyto waves triggered by fusion with the first sperm elicit immediate exocytosis of extracellular matrix-modifying enzymes to quickly block polyspermy15. In vitro fertilization studies in maize showed the formation of cell wall components already 30 s after gamete fusion60 indicating a quick release of cell wall components likely from trans-golgi vesicles stored in the egg cell. Whether this leads to a block of polyspermy, whether it also occurs in vivo and depends on Ca2+ signalling remains to be determined. However, the timing and the observation that central cell fusion lacks the second [Ca2+]cyto transient and allows polyspermy61 suggest that the egg cell-specific [Ca2+]cyto transient might also be involved to generate a quick block to polyspermy. In vitro fertilization studies using egg and central cells of tobacco confirm our finding that the second transient Ca2+ elevation is lacking in central cells62.
In conclusion, we have reported [Ca2+]cyto dynamics during the entire double fertilization process and were able to correlate the timing of Ca2+ signatures with cellular events including localized cross-talk between the pollen tube apex and synergid cells, pollen tube burst and sperm delivery, as well as gamete activation and fusion culminating in fertilization success and likely a polyspermy block to the egg cell. These studies thus significantly extend our knowledge about the timing of double fertilization events and deepen our understanding about the role of Ca2+ signalling during the individual processes. Future studies are necessary to unveil the molecular players involved in [Ca2+]cyto induction, signal maintenance and/or oscillation as well as the downstream targets regulating Ca2+ dependent processes to finally understand its role during double fertilization.
Methods
Generation of CerTN-L15 expressing plants
The coding sequence of CerTN-L15 (ref. 31) was amplified using the primers P12 (5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTGCCACCATG-3′) and P16 (5′-GGGGACCACTTTGTACAAGAAAGCTGGGTTCA-3′). The resulting PCR product was used in a BP-reaction (Gateway Technology manual; Life Technologies) with the donor vector pDONR207 (Invitrogen) resulting in pDONR207-CerTN-L15. SacI/SpeI restriction digest was performed to remove the 35S promoter of the destination vectors pB2GW7 and pH2GW7 (ref. 63), respectively, which was subsequently replaced by the promoters of either AtEC1.1 (ref. 12) (454 bp upstream of the start codon of At1g76750), AtMYB98 (ref. 33) (1.494 bp upstream of the start codon of At4g18770) or AtDD65 (ref. 34) (1.246 bp upstream of the start codon of At3g10890). The EC1.1 promoter was amplified using the primers P9 (5′-CAAAAGCTGGAGCTCTCTAGAG-3′) and P10 (5′-GGCCGACTAGTATCCTTCTCAAC-3′) from the plasmid pEC1.1-GUS12. The promoter pMYB98 was amplified with the primers P7 (5′-ATGGAGCTCGCGTCGACAGTTG-3′) and P8 (5′-AGGGACTGACTAGTGGATC-3′) from the plasmid pN7-Myb-EC1 (S. Sprunck, unpublished). pDD65 was amplified using the primers pDD65_FW (5′-AGTGAGCTCAATCAAAATTTAACATTTAAATAAAGTC-3′) and pDD65_REV (5′-TGATTACTAGTATCCTTTTCTACTTTGTTTTTGTTTTTGTGCT-3′) from Arabidopsis Col-0 genomic DNA (SacI and SpeI restriction sites underlined in primer sequences). CerTN-L15 (pDONR207-CerTN-L15) was recombined into the four destination vectors pB2GW7, pB2GW7-pEC1, pB2GW7-pMYB98 and pH2GW7-pDD65 by LR reaction (Gateway technology, Life Technologies) according to the manufacturer’s protocol. Resulting expression constructs (p35S::CerTN-L15, pEC1::CerTN-L15, pMYB98::CerTN-L15 and pDD65::CerTN-L15) were transformed into Arabidopsis Col-0 plants by floral dipping64. Transformants were selected according to their selection marker and fluorescent properties.
Generation of the double marker line LHR
The promoter pLat52 was amplified from Lat52::GFP expressing plants65 using the primers FV01 (5′-gggggagctcATACTCGACTCAGAAGGTATTGAGG-3′) and FV02 (5′-ggggactagttttaaattggaatttttttttttgg-3′) and cloned into the destination vector pB2GW7 (ref. 63) by replacing the 35S promoter using the introduced SacI and a SpeI sites, resulting in the destination vector pVOLat52GW. TagRFP was amplified from pTagRFP-C (Evrogen) with the primers ptagRFPfwd (5′-caccatggtgtctaagggcgaagagctg-3′) and ptagRFPrev (5′-tggctgattatgatcagttatctagatcc-3′) followed by insertion into the pENTR/D-TOPO vector according to the manufacturer’s protocol (Gateway Technology manual; Life Technologies) to obtain pENTR-tagRFP. To yield pENTR-tagRFP-T, site-directed mutagenesis was performed (QuikChange II Site-Directed Mutagenesis Kit, Agilent) using the primers tagRFP-TPOINT (5′-CTGGAAGGCAGAACCGACATGGCCCTG-3′) and TagRFP-TPOINTrev (5′-CAGGGCCATGTCGGTTCTGCCTTCCAG-3′). A C-terminal 109 nucleotide sequence of MtSYMREM1 (ref. 66), covering the C-terminal remorin anchor, was amplified from pDONR207-MtSYMREM1 with the primers FV03 (5′-GGGGAATTCTGGGGAAGAAATCCTCAAGGTAGAG-3′) and FV04 (5′-CCCCGGATCCCTAACTGAAAAACCTTAAACCGCTG-3′). The resulting PCR product was cloned into pENTR-tagRFP-T using the introduced EcoRI and BamHI restriction sites (underlined) to obtain pENTR-tagRFP-T-REM. The expression construct pPLat52:tagRFP-T-REM was created by LR reaction (Gateway Technology, Life Technologies) with pENTR-tagRFP-T-REM and pVOLat52GW. To create the double marker line Lat52:tagRFP-T-REM; HTR10:HTR10-mRFP (LHR), pPLat52:tagRFP-T-REM was used to transform plants homozygous for HTR10:HTR10-mRFP38 by floral dipping64. Transformants were selected according to their selection marker and fluorescent properties.
Growth conditions of Arabidopsis thaliana
Arabidopsis plants were either grown under short day conditions (8 h light, 20 °C, 50–70% relative humidity) for 4–6 weeks followed by long day conditions (16 h light, 20 °C, 50–70% relative humidity) or continuously grown under long day conditions.
Root imaging
Roots were prepared for imaging using the RootChip, a microfluidic perfusion device as described previously35,67. In brief, seeds were sterilized, placed on cut pipet tips (5 mm) that were filled with MS medium68 and incubated for 4 days under long day conditions in a growth chamber (16 h, 20 °C, 50–70% relative humidity). Seedlings were transferred to the RootChip and incubated for one additional day in liquid MS medium. Images were acquired every 3 s. Growing roots were continuously perfused with ½ strength MS medium and treated with square pulses of 200 mM mannitol over 5 min.
Semi-in vivo pollen tube growth assay
Semi-in vivo experiments were performed with modifications from the originally described assay32 using modified pollen germination medium (PGM)69 containing 18% sucrose, 0.01% boric acid, 1 mM MgSO4, 1 mM CaCl2, 1 mM Ca(NO3)2, 5 mM MES and 0.75% ultrapure agar (Version 1) or 0.175% agarose (peqGold Universal) (Version 2). The pH was adjusted to 7.0 with 200 mM KOH. The pistils and ovules that were used in all experiments were dissected from flowers with not yet opened anthers to ensure that only pollen used for pollination will fertilize the observed ovules. Preparation of pistils was performed by removing the leaves and anthers from the flower with forceps (Dumont No. 5) followed by cutting the pistil at junction of style and ovary with a razorblade and placing it upright on solidified PGM. Pollen was used from freshly opened anthers and put directly on the stigma of the cut pistils. For dissection of ovules, a pistil was cut open at the replum with the tip of a 27-gauge needle and the ovules were removed from the placenta using the needle. All dissections were performed on a microscope slide with double-sided tape to fix the tissue, but the contact of all tissue parts used for the assay with the adhesive tape was avoided.
Modified semi-in vivo assay
Version 1: A round plastic chamber (Ø1 × 1 cm), which was covered and sealed on one side with a microscope slide, was completely filled with 1 ml fresh PGM containing 0.75% agar. Medium was solidified for 30 min. The pollinated pistils and the individual ovules were placed on the medium surface. Afterwards, the chamber was covered by a cover slip with spacers of modelling clay, in a way that an air cushion was created between specimen and cover glass. The chamber was incubated at room temperature with light for 3 to 5 h. When the pollen tubes left the cut pistil and grew towards the dissected ovules, the chamber was turned cover glass-side down, the microscope slide was removed and the air cushion was released slowly, by puncturing the modelling clay with a needle. Thus, the agar helped to immobilize the tissue at the cover slip without disturbing the arrangement of the pistil and the ovules. After an additional incubation for 30 min, the chamber was used for imaging.
Version 2: All preparations were done under humidified conditions with a relative humidity of >80% by dissecting under a hood containing a humidifier. Pistils were cut and placed vertically on solid PGM containing 0.75% (w/v) agar. Stigmas of the pistils were pollinated and incubated for 3 h in the humidified chamber with light. Just before pollen tubes emerged from the pistil (usually ~3.5 h after pollination), pistils were transferred into a two-well microscope μSlide (IBIDI) that was filled with half solid PGM containing 0.175% (w/v) agarose. Before filling the μSlide with PGM, a 0.5-mm thick silicon mat with small slots, cut in the centre of the silicon layer, was placed on the bottom of the μSlide wells. Pistils were carefully pushed through the PGM to the bottom of the slide and fixed horizontally in the edges of the slots in the silicon layer. Afterwards, the ovules were dissected and transferred to the μSlide. They were pushed through the PGM with thin glass needles, pulled with a micropipette puller (P-97, Sutter Instruments), and placed in front of the cut pistils. The slides were covered with a lid and incubated in the humidified chamber with light. When pollen tubes had reached the ovules the μSlide was transferred to the microscope for imaging.
Microscopy
Imaging of the RootChip experiments was performed on an inverted epifluorescence microscope (LeicaDMIRE2) using a × 10 objective (Leica, 0.40 N.A., Air). For excitation of the sensor, a Polychrome V monochromator light source (TILL Photonics, Germany) was used. Fluorescence signals were detected using a Dual View beam splitter (Photometrics, USA) containing an ET470/24 m and ET535/30m filter setup and an EMCCD camera (QuantEM:512SC; Photometrics).
Semi-in vivo experiments were either done on an inverted Leica DMI6000B microscope using a × 40 objective (Leica, 1.25 N.A., Oil) equipped with a Yokogawa (Japan) CSU-X spinning disk confocal head and a motorized stage (ASI, USA) for automated multiposition imaging or on a Leica SP8 inverted confocal laser scanning microscope using a × 40 objective (Leica, 1.3 N.A, oil). Using the Leica DMI6000B microscope, fluorophores were excited at a wavelength of 441 nm (Cerulean), 514 nm (Citrine) and 561 (RFP/tagRFP) using a multiband dichroic mirror (445/515/561 nm dichroic beamsplitter, Semrock, USA) and the fluorescence signal was detected with an Evolve EMCCD camera (Photometrics, USA) using band-pass emission filters (475/42, 540/15 narrow, 605/64 RFP, Semrock). Exposure times were 100 ms (Citrine), 300 ms (Cerulean) and 600 ms (RFP). Acquisition was controlled using SlideBook 5.0 software (Intelligent Imaging Innovations, USA). Using the Leica SP8, fluorophores were excited with the same wavelengths described for the spinning disk system. Emission was detected using band-pass filter settings (Cerulean: 462–510 nm; Citrine: 520–550 nm and RFP/tagRFP: 570–666 nm).
Image processing and data analysis
Image processing and analysis were performed using FIJI ( http://fiji.sc/). To correct for movements or growth, images were registered using the StackReg plug-in ( http://bigwww.epfl.ch/thevenaz/stackreg/). To create false colour ratiometric images for FRET experiments, the RatioPlus plug-in ( http://rsbweb.nih.gov/ij/plugins/ratio-plus.html) and a 16-colour lookup table were used. For false colour ratiometric images as shown in Fig. 1b, raw data were processed as follows: multiplication of both ‘FRET’ channels (acceptor emission under donor excitation) with the reference channel (acceptor emission under acceptor excitation), 32-bit conversion, application of a mean filter with a radius of five pixels to both images, thresholding (Otsu-method) over the stack histogram. Dividing the acceptor channel by donor channel resulted in the ratiometric image (16-colour lookup table).
Analysis of FRET ratio changes was performed by calculating the ratio of fluorescence intensities of acceptor and donor under donor excitation using the MultiMeasure plug-in ( http://www.optinav.com/imagej.html) in specific regions of interest (Supplementary Fig. 6). The regions were 50 × 50 μm in root experiments and 4 × 4 μm squares for subcellular analysis or polygons in shape of the synergid cells for single cell analysis. The values of those measurements were further analysed in Excel. The ratios of acceptor and donor were normalized ((Ratio[t]−Ratio[t0])/Ratio[t0]) to display the relative ratio change over time. For the analysis of the [Ca2+]cyto oscillation in synergids, the individual 4 × 4 μm regions were aligned as described (Fig. 2d) and the regions were moved over time, following the position of the growing pollen tube tip. The amplitudes of the ratio changes in individual regions were calculated and the amplitude values of one oscillation event were normalized to the amplitude in region D1/L1. To define significant [Ca2+]cyto oscillations in synergid cells, a base line was determined as the mean ratio change over 10 min of FRET measurement before a pollen tube was in contact with the synergid cell. To determine the significance of amplitudes, the traces were smoothened applying a sliding average of three time points, the first derivative was calculated and the s.d. of the baseline (30 min without cell–cell interaction) of the resulting curve was determined. Amplitudes exceeding the s.d. of the baseline by fivefold (5 × σ) were considered significant.
Tracking of sperm cells was performed with the FIJI TrackMate plug-in ( http://fiji.sc/TrackMate) and resulting coordinates were plotted using excel. Kymographs for the analysis of sperm cell movement at the gamete fusion site were generated using the MultipleKymograph plug-in ( http://www.embl.de/eamnet/html/body_kymograph.html) along a segmented line that covered the track of each sperm cell separately. To compare the movement speed of the two sperm cells, the individual Kymographs were aligned with the starting point of the sperm cell movement overlaying with each other.
Additional Information
How to cite this article: Denninger, P. et al. Male–female communication triggers calcium signatures during fertilization in Arabidopsis. Nat. Commun. 5:4645 doi: 10.1038/ncomms5645 (2014).
References
Dresselhaus, T. & Franklin-Tong, N. Male-female cross-talk during pollen germination, tube growth and guidance, and double fertilization. Mol. Plant 6, 1018–1036 (2013).
Okuda, S. et al. Defensin-like polypeptide LUREs are pollen tube attractants secreted from synergid cells. Nature 458, 357–361 (2009).
Márton, M. L., Cordts, S., Broadhvest, J. & Dresselhaus, T. Micropylar pollen tube guidance by egg apparatus 1 of maize. Science 307, 573–576 (2005).
Takeuchi, H. & Higashiyama, T. A species-specific cluster of defensin-like genes encodes diffusible pollen tube attractants in Arabidopsis. PLoS Biol. 10, e1001449 (2012).
Márton, M. L., Fastner, A., Uebler, S. & Dresselhaus, T. Overcoming hybridization barriers by the secretion of the maize pollen tube attractant ZmEA1 from Arabidopsis ovules. Curr. Biol. 22, 1194–1198 (2012).
Capron, A. et al. Maternal control of male-gamete delivery in Arabidopsis involves a putative GPI-anchored protein encoded by the LORELEI gene. Plant Cell 20, 3038–3049 (2008).
Escobar-Restrepo, J.-M. et al. The FERONIA receptor-like kinase mediates male-female interactions during pollen tube reception. Science 317, 656–660 (2007).
Kessler, S. A. et al. Conserved molecular components for pollen tube reception and fungal invasion. Science 330, 968–971 (2010).
Amien, S. et al. Defensin-like ZmES4 mediates pollen tube burst in maize via opening of the potassium channel KZM1. PLoS Biol. 8, e1000388 (2010).
Woriedh, M. et al. External application of gametophyte-specific ZmPMEI1 induces pollen tube burst in maize. Plant Reprod. 26, 255–266 (2013).
Hamamura, Y. et al. Live-cell imaging reveals the dynamics of two sperm cells during double fertilization in Arabidopsis thaliana. Curr. Biol. 21, 497–502 (2011).
Sprunck, S. et al. Egg cell-secreted EC1 triggers sperm cell activation during double fertilization. Science 338, 1093–1097 (2012).
Iwao, Y. Egg activation in physiological polyspermy. Reproduction 144, 11–22 (2012).
Gilkey, J. C., Jaffe, L. F., Ridgway, E. B. & Reynolds, G. T. A free calcium wave traverses the activating egg of the medaka, Oryzias latipes. J. Cell Biol. 76, 448–466 (1978).
Miao, Y.-L. et al. Calcium influx-mediated signaling is required for complete mouse egg activation. Proc. Natl Acad. Sci. USA 109, 4169–4174 (2012).
Ma, W. & Berkowitz, G. A. The grateful dead: calcium and cell death in plant innate immunity. Cell. Microbiol. 9, 2571–2585 (2007).
Oldroyd, G. E. D., Harrison, M. J. & Paszkowski, U. Reprogramming plant cells for endosymbiosis. Science 324, 753–754 (2009).
Holdaway-Clarke, T. L., Feijo, J. A., Hackett, G. R., Kunkel, J. G. & Hepler, P. K. Pollen tube growth and the intracellular cytosolic calcium gradient oscillate in phase while extracellular calcium influx is delayed. Plant Cell 9, 1999–2010 (1997).
Schiøtt, M. et al. A plant plasma membrane Ca2+ pump is required for normal pollen tube growth and fertilization. Proc. Natl Acad. Sci. USA 101, 9502–9507 (2004).
Iwano, M. et al. Cytoplasmic Ca2+ changes dynamically during the interaction of the pollen tube with synergid cells. Development 139, 4202–4209 (2012).
Boisson-Dernier, A. et al. ANXUR receptor-like kinases coordinate cell wall integrity with growth at the pollen tube tip via NADPH oxidases. PLoS Biol. 11, e1001719 (2013).
Faure, J. E., Digonnet, C. & Dumas, C. An in vitro system for adhesion and fusion of maize gametes. Science 263, 1598–1600 (1994).
Antoine, A. F. et al. A calcium influx is triggered and propagates in the zygote as a wavefront during in vitro fertilization of flowering plants. Proc. Natl Acad. Sci. USA 97, 10643–10648 (2000).
Antoine, A. F., Faure, J. E., Dumas, C. & Feijo, J. A. Differential contribution of cytoplasmic Ca2+ and Ca2+ influx to gamete fusion and egg activation in maize. Nat. Cell Biol. 3, 1120–1123 (2001).
Digonnet, C., Aldon, D., Leduc, N., Dumas, C. & Rougier, M. First evidence of a calcium transient in flowering plants at fertilization. Development 124, 2867–2874 (1997).
Miyawaki, A. et al. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 388, 882–887 (1997).
Nagai, T., Yamada, S., Tominaga, T., Ichikawa, M. & Miyawaki, A. Expanded dynamic range of fluorescent indicators for Ca(2+) by circularly permuted yellow fluorescent proteins. Proc. Natl Acad. Sci. USA 101, 10554–10559 (2004).
Monshausen, G. B., Messerli, M. A. & Gilroy, S. Imaging of the Yellow Cameleon 3.6 indicator reveals that elevations in cytosolic Ca2+ follow oscillating increases in growth in root hairs of Arabidopsis. Plant Physiol. 147, 1690–1698 (2008).
Krebs, M. et al. FRET-based genetically encoded sensors allow high-resolution live cell imaging of Ca(2+) dynamics. Plant J. 69, 181–192 (2012).
Heim, N. & Griesbeck, O. Genetically encoded indicators of cellular calcium dynamics based on troponin C and green fluorescent protein. J. Biol. Chem. 279, 14280–14286 (2004).
Heim, N. et al. Improved calcium imaging in transgenic mice expressing a troponin C–based biosensor. Nat. Methods 4, 127–129 (2007).
Palanivelu, R. & Preuss, D. Distinct short-range ovule signals attract or repel Arabidopsis thaliana pollen tubes in vitro. BMC Plant Biol. 6, 7 (2006).
Kasahara, R. D., Portereiko, M. F., Sandaklie-Nikolova, L., Rabiger, D. S. & Drews, G. N. MYB98 is required for pollen tube guidance and synergid cell differentiation in Arabidopsis. Plant Cell 17, 2981–2992 (2005).
Steffen, J. G., Kang, I.-H., Macfarlane, J. & Drews, G. N. Identification of genes expressed in the Arabidopsis female gametophyte. Plant J. 51, 281–292 (2007).
Grossmann, G. et al. The RootChip: an integrated microfluidic chip for plant science. Plant Cell 23, 4234–4240 (2011).
Kiegle, E., Moore, C. A., Haseloff, J., Tester, M. A. & Knight, M. R. Cell-type-specific calcium responses to drought, salt and cold in the Arabidopsis root. Plant J. 23, 267–278 (2000).
Leshem, Y., Johnson, C. & Sundaresan, V. Pollen tube entry into the synergid cell of Arabidopsis is observed at a site distinct from the filiform apparatus. Plant Reprod. 26, 93–99 (2013).
Ingouff, M., Hamamura, Y., Gourgues, M., Higashiyama, T. & Berger, F. Distinct dynamics of HISTONE3 variants between the two fertilization products in plants. Curr. Biol. 17, 1032–1037 (2007).
Garaschuk, O., Griesbeck, O. & Konnerth, A. Troponin C-based biosensors: a new family of genetically encoded indicators for in vivo calcium imaging in the nervous system. Cell Calcium 42, 351–361 (2007).
Kaestner, L. Calcium Signalling Springer (2012).
Ngo, Q. A., Vogler, H., Lituiev, D. S., Nestorova, A. & Grossniklaus, U. A calcium dialog mediated by the FERONIA signal transduction pathway controls plant sperm delivery. Dev. Cell. 29, 491–500 (2014).
Dodd, A. N., Kudla, J. & Sanders, D. The language of calcium signaling. Annu. Rev. Plant Biol. 61, 593–620 (2010).
Meyer, T. & Stryer, L. Molecular model for receptor-stimulated calcium spiking. Proc. Natl Acad. Sci. USA 85, 5051–5055 (1988).
Ehrhardt, D. W., Wais, R. & Long, S. R. Calcium spiking in plant root hairs responding to Rhizobium nodulation signals. Cell 85, 673–681 (1996).
Chabaud, M. et al. Arbuscular mycorrhizal hyphopodia and germinated spore exudates trigger Ca2+ spiking in the legume and nonlegume root epidermis. New Phytol. 189, 347–355 (2011).
Kosuta, S. et al. Differential and chaotic calcium signatures in the symbiosis signaling pathway of legumes. Proc. Natl Acad. Sci. USA 105, 9823–9828 (2008).
Oldroyd, G. E. D. & Downie, J. A. Coordinating nodule morphogenesis with rhizobial infection in legumes. Annu. Rev. Plant Biol. 59, 519–546 (2008).
Monshausen, G. B., Bibikova, T. N., Weisenseel, M. H. & Gilroy, S. Ca2+ regulates reactive oxygen species production and pH during mechanosensing in Arabidopsis roots. Plant Cell 21, 2341–2356 (2009).
Nakagawa, Y. et al. Arabidopsis plasma membrane protein crucial for Ca2+ influx and touch sensing in roots. Proc. Natl Acad. Sci. USA 104, 3639–3644 (2007).
Uhlén, P. & Fritz, N. Biochemistry of calcium oscillations. Biochem. Biophys. Res. Commun. 396, 28–32 (2010).
Wilkins, K. A., Poulter, N. S. & Franklin-Tong, V. E. Taking one for the team: self-recognition and cell suicide in pollen. J. Exp. Bot. 65, 1331–1342 (2014).
Beale, K. M., Leydon, A. R. & Johnson, M. A. Gamete fusion is required to block multiple pollen tubes from entering an Arabidopsis ovule. Curr. Biol. 22, 1090–1094 (2012).
Völz, R., Heydlauff, J., Ripper, D., Lyncker, von, L. & Gross-Hardt, R. Ethylene signaling is required for synergid degeneration and the establishment of a pollen tube block. Dev. Cell 25, 310–316 (2013).
Orrenius, S., Zhivotovsky, B. & Nicotera, P. Regulation of cell death: the calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 4, 552–565 (2003).
Duan, Q. et al. Reactive oxygen species mediate pollen tube rupture to release sperm for fertilization in Arabidopsis. Nat. Commun. 5, 3129 (2014).
Haruta, M., Sabat, G., Stecker, K., Minkoff, B. B. & Sussman, M. R. A peptide hormone and its receptor protein kinase regulate plant cell expansion. Science 343, 408–411 (2014).
Swann, K. & Yu, Y. The dynamics of calcium oscillations that activate mammalian eggs. Int. J. Dev. Biol. 52, 585–594 (2008).
Hamamura, Y. et al. Calcium spikes during double fertilization in Arabidopsis. Nat. Commun. 5, 4722 (2014).
Okabe, M. The cell biology of mammalian fertilization. Development 140, 4471–4479 (2013).
Kranz, E., Wiegen, P. & Lörz, H. Early cytological events after induction of cell division in egg cells and zygote development following in vitro fertilization with angiosperm gametes. Plant J. 8, 9–23 (1995).
Scott, R. J., Armstrong, S. J., Doughty, J. & Spielman, M. Double fertilization in Arabidopsis thaliana involves a polyspermy block on the egg but not the central cell. Mol. Plant 1, 611–619 (2008).
Peng, X.-B., Sun, M.-X. & Yang, H.-Y. Comparative detection of calcium fluctuations in single female sex cells of tobacco to distinguish calcium signals triggered by in vitro fertilization. J. Integr. Plant Biol. 51, 782–791 (2009).
Karimi, M., Depicker, A. & Hilson, P. Recombinational cloning with plant gateway vectors. Plant Physiol. 145, 1144–1154 (2007).
Clough, S. J. & Bent, A. F. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743 (1998).
Twell, D., Yamaguchi, J. & McCormick, S. Pollen-specific gene expression in transgenic plants: coordinate regulation of two different tomato gene promoters during microsporogenesis. Development 109, 705–713 (1990).
Konrad, S. S. A. et al. S-acylation anchors Remorin proteins to the plasma membrane but does not primarily determine their localization in membrane micro-domains. New Phytol. 203, 758–769 (2014).
Grossmann, G. et al. Time-lapse fluorescence imaging of Arabidopsis root growth with rapid manipulation of the root environment using the RootChip. J. Vis. Exp. 65, 4290 (2012).
Murashige, T. & Skoog, F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol. Plant 15, 473–497 (1962).
Li, H., Lin, Y., Heath, R. M., Zhu, M. X. & Yang, Z. Control of pollen tube tip growth by a Rop GTPase-dependent pathway that leads to tip-localized calcium influx. Plant Cell 11, 1731–1742 (1999).
Acknowledgements
We wish to thank Oliver Griesbeck for the CerTN-L15 construct, Fred Berger for the HTR10-mRFP marker line and Sheila McCormick for Lat52:GFP expressing plants. We are grateful to Heather Cartwright for help with imaging, to Birgit Absmanner for assistance with cloning and Ulrich Hammes for stimulating discussions. Financial support by the German Research Foundation DFG (grant SP 686/1–2 to S.S., grant SFB924 to T.D., T.O. and S.S. and SFB960 to T.D.), the German Academic Exchange Service DAAD (to P.D.), an Emmy-Noether grant of the DFG (to T.O.), an EMBO long-term fellowship and the German Excellence Initiative (to G.G.), grants from the Department of Energy (DE-FG02-04ER15542; to W.B.F.) and funds from the Carnegie Institution for Science (to D.W.E., W.B.F. and G.G.) German Excellence Initiative (EXC81; to G.G.), are gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
T.D. conceived the project; T.D., W.B.F. and G.G. planned the experiments; P.D. and A.B. conducted the experiments; A.B., A.L. and F.V. created transgenic plant lines; S.S. and T.O. contributed unpublished materials; P.D. and G.G. performed data analyses; T.D., G.G., S.S. and D.W.E. supervised the project; P.D. and G.G. prepared figures and Supplementary information; G.G., T.D. and P.D. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-6 (PDF 1933 kb)
Supplementary Movie 1
Ca2+ cyto oscillations during the contact of synergid cells with a pollen tube. Time series corresponding to Figure 1b. Left panel: YFP signal of pMyb98::CerTN-L15 in yellow, signal of LHR pollen tube membrane and sperm nuclei in red. Right panel: corresponding Ca2+ cyto level in the synergid cells shown as ratiometric images of pMyb98::CerTN-L15. Time interval, 10 sec. (AVI 6085 kb)
Supplementary Movie 2
Localized Ca2+ cyto oscillations in synergid cells in contact with the pollen tube apex. Time series of CerTN-L15 response shown in false colour and merged with LHR signals (grey scale). Time interval, 10 sec. (AVI 2090 kb)
Supplementary Movie 3
Fertilization of a pDD65::CerTN-L15 ovule (yellow central cell). Time series corresponds to Graph 3 in Supplementary Figure 3a. YFP signal of pDD65::CerTN-L15 shown in yellow, sperm cell nuclei of LHR pollen in red. Time interval, 10 sec. (AVI 7625 kb)
Supplementary Movie 4
Ca2+ cyto signature of an egg cell during fertilization. Time series corresponding to egg cell shown in Figure 3c and calcium recording shown in graph 1 in 4c. Left panel: YFP signal of pEC1::CerTN-L15 shown in yellow, sperm nuclei in red. Right panel: Ratiometric image series of CerTN-L15 merged with LHR signal (grey scale). Time interval, 10 sec. (AVI 4749 kb)
Supplementary Movie 5
Unsuccessful fertilization attempt of the egg cell and one sperm cell nucleus. Time series corresponding to Supplementary Figures 4a,b. Time interval, 10 sec. (AVI 4960 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Denninger, P., Bleckmann, A., Lausser, A. et al. Male–female communication triggers calcium signatures during fertilization in Arabidopsis. Nat Commun 5, 4645 (2014). https://doi.org/10.1038/ncomms5645
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms5645
This article is cited by
-
EGG CELL 1 contributes to egg-cell-dependent preferential fertilization in Arabidopsis
Nature Plants (2024)
-
Distribution of exchangeable Ca2+ during the process of Larix decidua Mill. pollination and germination
Scientific Reports (2024)
-
Synergid cell calcium oscillations refine understanding of FERONIA/LORELEI signaling during interspecific hybridization
Plant Reproduction (2024)
-
RALF signaling pathway activates MLO calcium channels to maintain pollen tube integrity
Cell Research (2023)
-
Rapid alkalinization factor: function, regulation, and potential applications in agriculture
Stress Biology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
