
Abstract
Current interferon alpha-based treatment of hepatitis C virus (HCV) infection fails to cure a sizeable fraction of patients treated. The cause of this treatment failure remains unknown. Here using mathematical modelling, we predict treatment failure to be a consequence of the emergent properties of the interferon-signalling network. HCV induces bistability in the network, creating a new steady state where it can persist. Cells that admit the new steady state alone are refractory to interferon. Using a model of viral kinetics, we show that when the fraction of cells refractory to interferon in a patient exceeds a critical value, treatment fails. Direct-acting antivirals that suppress HCV replication can eliminate the new steady state, restoring interferon sensitivity and improving treatment response. Our study thus presents a new conceptual basis of HCV persistence and treatment response, elucidates the origin of the synergy between interferon and direct-acting antivirals, and facilitates rational treatment optimization.
Similar content being viewed by others
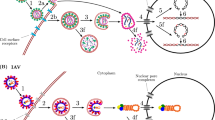

Introduction
Hepatitis C virus (HCV) infection is spread over 130–170 million people worldwide1. Pegylated interferon-α (IFN-α), in combination with ribavirin, forms the mainstay of the current treatment for HCV infection. The combination (PR), however, elicits a sustained virological response (SVR) in only about 50% of HCV genotype 1 infected patients treated2. Several factors that correlate with treatment response have been identified, including HCV genotype, pre-treatment viral load and single-nucleotide polymorphisms (SNPs) in the IFN-λ locus3,4. The cause of treatment failure, however, remains poorly established.
IFN-α belongs to a family of cytokines produced endogenously in response to viral infection5,6. Through a complex series of signalling events involving the JAK–STAT pathway, stimulation by IFN results in the expression of several hundred IFN-stimulated genes (ISGs) which together induce an antiviral state in cells and control infection6. With HCV, however, a vast majority (~70–80%) of infected individuals are unable to clear the infection spontaneously and become chronically infected6,7. PR treatment can compensate for suboptimal immune control of infection. ISG expression following the onset of treatment was enhanced substantially over the pre-treatment level in the livers of HCV patients who responded to PR8,9,10. (Ribavirin alone elicits no SVR11, but improves response in combination with IFN12, suggesting that ribavirin potentiates the anti-HCV activity of IFN10,13,14,15.) Surprisingly, however, in the livers of nonresponders to PR treatment, pre-treatment ISG expression was higher than in responders and did not increase significantly following the onset of treatment8,9,10.
Non-responsiveness to PR was eliminated in part by the recently approved direct-acting antiviral agents (DAAs) telaprevir and boceprevir, each of which in conjunction with PR yielded SVR rates of over 70% in previously untreated HCV genotype 1 infected patients2,16. Because telaprevir and boceprevir monotherapy fail owing to the emergence of drug resistance, one hypothesis for the success of the triple combination is that DAAs act on drug sensitive strains whereas PR control resistant strains17. Paradoxically, however, although the DAAs alone elicited no SVR, the triple combination induced SVR in 40% of previous null responders to PR, who showed no significant decline in viral levels during PR treatment indicating the near absence of IFN activity18,19. DAAs thus appear to synergize with IFN and help overcome non-responsiveness to PR.
In two recent pioneering studies, poor response following high-pre-treatment ISG expression has been suggested to arise from upregulation, associated with persistent IFN signalling, of immunosuppressive molecules such as interleukin-10 and programmed death-ligand 1 (PD-L1), which leads to T-cell exhaustion and viral persistence20,21. These studies mark a breakthrough in our understanding of chronic viral infections and recommend, counter intuitively, suppression of IFN signalling to avert T-cell exhaustion and elicit control of chronic infection. Two key questions, however, remain unanswered. When does HCV infection lead to high (pre-treatment) ISG expression and therefore poor-treatment response? How, despite high-exogenous IFN addition, which would ensure continued immunosuppression following the onset of treatment, do DAAs improve treatment response in combination with PR? Mechanisms in addition to IFN-associated immunosuppression thus appear to contribute to HCV persistence and treatment response.
Previous studies have identified several mechanisms by which HCV evades IFN activity: HCV can suppress endogenous IFN production, inhibit ISG expression by interfering with the JAK–STAT pathway, induce a block in the translation of ISG mRNA, and/or inhibit ISG effector functions6,22. At the same time, key ISGs involved in the control of HCV have been identified and in some cases their mode of action elucidated23,24,25,26,27. These specific interactions between HCV and components of the IFN network have unravelled the many fronts of the battle between HCV and our innate immune system. The outcome of this battle, however, has been difficult to predict: the specific interactions prove inadequate for distinguishing between nonresponders and responders to PR or explaining the observed synergy between DAAs and PR, precluding rational strategies for improving treatment response.
In the present study, we explore the possibility that responsiveness to IFN is not a consequence of specific molecular interactions between individual components of the IFN system and HCV alone, but of emergent, systems-level properties of the IFN signalling network interacting with HCV. The IFN network is designed to suppress HCV upon stimulation. HCV, on the other hand, interferes with IFN signalling and compromises its effector functions. Non-responsiveness to IFN may thus be a manifestation of the state to which the IFN system is driven as a consequence of these competing interactions. Further, although DAAs directly target viral (or host) proteins, their action may, at the systems level, weaken the influence of HCV on the IFN network and drive the system to an alternative, responsive state. To test this hypothesis, we construct a mathematical model of the IFN signalling network in the presence of HCV and elucidate its systems-level properties. We find intriguingly that HCV induces bistability in the network, causing the emergence of a new steady state where HCV persists. Accounting for this bistability, we develop a mathematical model of viral kinetics and deduce a criterion for treatment response. Finally, we find that DAAs can eliminate this bistability and improve treatment response.
Results
Model of the IFN signalling network in the presence of HCV
We considered the intracellular IFN signalling network in the presence of HCV (Fig. 1). Upon infection, HCV replicates inside the cell producing copies of positive and negative strands of RNA, the latter presumably existing as double-stranded RNA (dsRNA). In the presence of IFN, ISGs are transcribed to produce ISG mRNA. The translation of ISG mRNA yields ISG proteins, which control HCV by inhibiting different steps of the HCV life cycle and/or lowering the stability of HCV RNA23,24,25,26,27. Of the ninety ISGs expressed in response to IFN (type I) stimulation, seven were recently found to exhibit significant activity against HCV24. HCV can evade IFN action by several mechanisms including lowering global translation in the cell via protein kinase R (PKR)28. The latter mechanism has been shown in recent in vitro studies to facilitate HCV persistence despite strong IFN treatment28, postulated to be the predominant mode of the interference of IFN signalling by HCV in vivo29, and may represent a key pathway underlying the resistance of HCV to IFN30. We therefore focused here on the HCV-mediated block in the translation of ISG mRNA via PKR. (We also considered other mechanisms of the interference of IFN signalling by HCV and found similar results; see Discussion.) Initiation of translation depends on the abundance of the eIF2α–guanosine triphosphate (GTP) complex. eIF2α–GTP gets hydrolyzed to eIF2α–guanosine diphosphate (GDP) during translation31. The recycling of eIF2α–GDP to eIF2α–GTP is mediated by another protein eIF2B32. HCV induces PKR dimerization, presumably by the binding of HCV double-stranded RNA to PKR33,34. PKR dimers undergo autophosphorylation34. Phosphorylated PKR catalyzes the phosphorylation of eIF2α–GDP. Phosphorylated eIF2α–GDP does not undergo GDP to GTP exchange and has an increased affinity for eIF2B31,32. eIF2B is present in low abundance compared with eIF2α31. Thus, phosphorylated eIF2α–GDP sequesters eIF2B, which further reduces the eIF2α–GTP level, blocking translation. HCV, however, continues to replicate as its translation is less sensitive to eIF2α–GTP28,35,36,37. The HCV–PKR interaction thus contains key elements of the suppression of IFN activity by HCV as well as the control of HCV by IFN, capturing the essence of the battle between HCV and IFN at the systems level. We modelled the network using a system of coupled differential equations (Supplementary Note 1) and elucidated its emergent properties.

Following exposure to IFN, signalling via the JAK–STAT pathway triggers ISG expression, which lowers the HCV RNA level inside a cell. HCV catalyzes PKR dimerization and autophosphorylation. Phosphorylated PKR phosphorylates eIF2α-GDP, which sequesters eIF2B and lowers translation of ISG mRNA. Inset: schematic of the double-negative feedback in the network owing to the suppression of HCV by ISG proteins and of ISG proteins by HCV.
Bistability in the IFN signalling network induced by HCV
The network exhibits double-negative feedback: ISG proteins control HCV, whereas HCV controls ISG mRNA translation. Double-negative feedback with ultra-sensitivity (nonlinearity) can give rise to bistability38, observed in several signalling and transcription networks38,39,40,41,42. To test the existence of bistability, we identified the steady states of the network as follows. We predicted first the dependence of the steady state ISG protein level on the HCV RNA level (red line in Fig. 2). Here the double-stranded HCV RNA level was fixed, thus eliminating the negative feedback of ISG on HCV, and the system solved for steady state. This process was repeated for different values of the double-stranded HCV RNA level. Similarly, we predicted the dependence of the steady state HCV RNA level on the ISG protein level (blue line in Fig. 2), by suppressing the negative feedback of HCV on ISG. When both the feedback loops are active, the intersection points Q1, Q2 and Q3 represent the steady states of the system (Fig. 2). At Q1, the HCV RNA level is high and the ISG protein level low, marking HCV persistence, whereas at Q3, HCV RNA is absent and the ISG protein level is high, indicating HCV clearance. Q1 and Q3 are the two stable steady states of the system. Q2, where HCV RNA and ISG protein levels are intermediate, is an unstable steady state, in that small perturbations get amplified and drive the system away from Q2. Q2, thus acts as a threshold determining which of the two stable steady states the system eventually attains.

Model predictions of concentration-effect curves showing steady state positive-strand HCV RNA levels for fixed total ISG protein levels (blue line) and total ISG protein levels for fixed double-stranded HCV RNA levels (red line). Both curves are drawn with positive-strand HCV RNA levels on the y axis and ISG protein levels on the x axis. The ISG proteins include the seven ISGs, namely, IFIT3, IFITM1, IFITM3, PLSCR1, TRIM14, Viperin and RNase L, identified recently to have anti-HCV activity24 (Supplementary Note 1). The intersections of the curves mark the three steady states, Q1, Q2 and Q3, of the network. Parameters employed are listed in Supplementary Tables 1–3.
Bistability in in vitro experiments and model validation
Evidence of this threshold and the underlying bistability is found in recent in vitro experiments28. In the latter experiments, cells were treated with IFN before or at different times post infection and HCV RNA and ISG protein levels were measured 20 h post IFN treatment28. When treated up to 24 h post infection, phosphorylated PKR levels were low, ISG protein levels were high and HCV RNA levels suppressed. When treated ≥48 h post infection, phosphorylated PKR levels were high, ISG protein levels low and HCV RNA levels approached that of IFN untreated cells. Using parameter values obtained independently (Supplementary Note 2, Supplementary Tables 1–3), our model captured the above observations quantitatively (Fig. 3a). According to our model, following infection, the HCV RNA level gradually increases inside cells (Supplementary Fig. 1A); endogenous IFN production is not expected in the in vitro cell-culture infection system43. If IFN is added when the HCV RNA level is below the threshold corresponding to Q2, the system drops to Q3, where HCV is controlled. In contrast, if IFN is added after the HCV RNA level crosses the threshold, the system reaches Q1, where HCV persists. The switch in the response of the system based on the time of IFN addition is thus a signature of bistability. Indeed, silencing PKR, which suppresses one of the feedback loops, eliminated this signature in the experiments and in our model predictions (Fig. 3b). Further, our model mimicked all the observations of the latter in vitro study28 (Supplementary Note 3, Supplementary Figs 1–3), giving us confidence in its ability to accurately unravel the behaviour of the underlying network.

Normalized positive-strand HCV RNA levels predicted by our model (orange) and measured previously in in vitro experiments28 (green) at 20 h post IFN treatment (100 IU ml−1) following different specified times (hours post infection) with HCV. Error bars represent standard deviations from triplicates. Lower inset: model predictions of corresponding HCV RNA levels achieved at steady state post IFN treatment. Upper inset: model predictions of corresponding ISG protein levels at 20 h (orange) and at steady state (blue) post IFN treatment. (a) normal cells and (b), PKR-silenced cells (60% reduction in PKR mRNA transcription rate). The dashed line marks zero. Normalization was done with the positive-strand HCV RNA level in cells without IFN treatment. Parameters employed are listed in Supplementary Tables 1–3.
Diversity of the cellular response to IFN
We found that the network exhibited bistability over several-fold changes in the values of several parameters (Supplementary Fig. 4). Global sensitivity analysis yielded parameters to which the systems-level properties of the network were sensitive (Supplementary Note 4, Supplementary Fig. 4). Bistability was lost when parameter values tilted the competition between HCV and IFN heavily in favour of either one. For instance, when the repression coefficients for the control of HCV replication by ISGs were decreased, IFN effectiveness increased and the system admitted the steady state Q3 alone, where HCV is eliminated. When the repression coefficients were increased, IFN effectiveness decreased and the system admitted Q1 alone, where HCV thrives (Fig. 4a).

Steady states of the network, Q1 (pink), Q2 (cyan) and Q3 (green) over a range of values of (a), the repression coefficients for the control of HCV replication by ISGs (ω denotes the fold-change in the repression coefficients of all ISGs relative to their baseline values in Supplementary Table 3), and (b), the IFN level. The three distinct cellular response phenotypes (see text) are marked. Region I admits Q1 alone, region II both Q1 and Q3 and region III Q3 alone. In b, region III does not occur within the range of IFN levels considered. Other parameters employed are listed in Supplementary Tables 1–3.
It follows that intrinsic variations across cells, for example, in ISG expression44,45 or the expression of host protease and other enzymes that affect HCV replication, would yield three distinct cellular phenotypes based on the response to a given level of IFN stimulation, viz., cells that admit Q1 alone, Q3 alone or both Q1 and Q3. Cells that admit Q1 alone would allow HCV persistence even with IFN stimulation. Cells that admit Q3 alone would clear HCV following IFN stimulation. Cells that admit both Q1 and Q3 would either allow HCV persistence or clear HCV depending on the time of IFN addition, as illustrated above. Increasing the level of IFN stimulation could convert a cell that admits Q1 alone to one that admits both Q1 and Q3 and further to one that admits Q3 alone. Decreasing the level of IFN stimulation could reverse these transitions. Bistable systems, however, display hysteretic behaviour: the level of IFN stimulation required to drive a cell from Q1 to Q3 would be larger than the level at which the cell transitions from Q3 to Q1. The former level may lie beyond experimentally/clinically employed levels of IFN28 (Fig. 4b). Thus, cells in the state Q1 may allow HCV persistence despite seemingly high levels of IFN stimulation and are therefore predicted to be refractory to IFN. In contrast, cells in the state Q3 clear HCV infection with much lower levels of IFN stimulation following exposure to HCV (Fig. 4b). The latter cells are thus sensitive to IFN. How an HCV-infected individual responds to IFN-based treatment would thus be determined by the distribution of cells in the individual across the different IFN response phenotypes.
Viral kinetics and treatment outcome
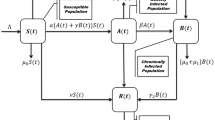
To predict the ensuing viral kinetics and treatment outcome, we advanced the basic model of viral kinetics in vivo14,46,47,48,49 (Supplementary Note 5) by explicitly considering cell subpopulations with the three distinct IFN response phenotypes (Fig. 5a). We divided target cells into subpopulations Ti with i=1, 2 and 3 representing cells that admit Q1 alone, both Q1 and Q3, and Q3 alone, respectively, at the level of IFN exposure employed in the treatment; because ribavirin potentiates IFN activity, the subpopulations may be defined based on the combined activity of PR. Correspondingly, infected cells are divided into subpopulations I1, I2 and I3. From the above analysis, it follows that cells T1 remain susceptible to infection and are refractory to IFN. IFN does not inhibit de novo infection in this subpopulation. Cells T2 are sensitive to IFN, as IFN exposure before HCV infection drives these cells to the state Q3 and prevents de novo infections. Cells T3 are similarly protected from new infections. Cells I1 are again refractory to IFN and continue viral production. Cells I2 are infected before IFN exposure and thus attain the state Q1 following IFN exposure and continue to produce virions. Cells I3 admit Q3 alone and are cured following exposure to IFN.

(a) Schematic of viral kinetics incorporating distinct cellular responses to IFN. Top: cells that have poor IFN control of HCV (left) admit Q1 alone and are refractory to IFN (right). Middle: cells that have intermediate IFN control of HCV and vice versa (left) admit both Q1 and Q3 and thus continue viral production when infected but are protected from new infections by IFN (right). Bottom: cells that have strong IFN control of HCV (left) admit Q3 alone and are sensitive to IFN (right). Target cells in the three subpopulations, Ti, where i=1, 2 and 3, are produced at the rates si, proliferate with the rate constant rT, die with the rate constant dT and are lost owing to de novo infection with the rate constant β. The corresponding infected cells Ii proliferate with the rate constant rI and die with the rate constant δ. Free virions are produced by infected cells with the rate constant p and are cleared with the rate constant c. The de novo infection rate and virion production rate were assumed to be blocked with efficacies ηi and εi in the respective subpopulations owing to PR treatment. (b) Model predictions of different viral response patterns following IFN treatment for different distributions of cells in the three IFN response phenotypes: φ1=1, φ2=0, and φ3=0 (green top); φ1=0.35, φ2=0.35, and φ3=0.3 (green bottom); φ1=0.015, φ2=0.185, and φ3=0.8 (red); and φ1=0, φ2=0.2, and φ3=0.8 (blue). (c) Steady state viral levels following IFN treatment as a function of φ1 when φ2=φ3 (solid line) and φ2=0.5φ3 (dashed line) (the two curves overlap) and (Inset) long-term virological response corresponding to the predictions in b. Parameters: V0=7 × 106 IU ml−1, β=3 × 10−7 ml per virions per day. Other parameter values employed are listed in Supplementary Table 4.
The resulting viral kinetics typically follows a biphasic decline (Fig. 5b). We estimate with our model that the viral load at the end of the first phase is ~V0(1–φ3), where V0 is the baseline viral load and φ3 is the pre-treatment frequency of IFN-sensitive cells (Supplementary Note 6, Supplementary Fig. 5A). The extent of the first phase decline is thus determined by the fraction of cells cured by IFN. The second phase slope depends on the dynamics of the infected cells. Whereas cells I2 and I3 decline at the death rate δ—de novo infection is blocked by IFN in these subpopulations and infected cells are expected not to proliferate significantly48 (Supplementary Table 4)—the loss of cells I1 is compensated at least in part by the de novo infection of cells T1, refractory to IFN (Supplementary Note 5, Supplementary Fig. 5B). The early second phase slope estimated using our model is ~δ(1–φ1). The second phase slope thus decreases as the pre-treatment frequency of IFN-refractory cells, φ1, increases (Supplementary Note 6, Supplementary Fig. 5C). By varying the frequencies φ1, φ2 and φ3, our model reproduced all the patterns of viral kinetics observed in patients (Fig. 5b).
The second phase slope in our model depends on φ1. Indeed, we find that when φ1 is below a critical value,  , viral clearance is achieved (Fig. 5c). This critical value does not depend on φ2 and φ3 because following treatment, cells T2 and T3 do not acquire new infections and cells I2 and I3 are eventually lost owing to cell death even when IFN fails to cure them. IFN-refractory cells, T1, remain susceptible to new infections even during treatment and thus determine treatment outcome. When their number is sufficiently small, viral production from infected cells is inadequate to replenish the loss of infected cells owing to cell death (akin to the basic reproductive number being below unity50), resulting in the eventual clearance of infection. Thus, the success of IFN treatment depends primarily on the size of the IFN-refractory subpopulation. The larger the latter subpopulation, the smaller the second phase slope, thus presenting an explanation of the strong correlation observed between the second phase slope and treatment response46,51,52.
, viral clearance is achieved (Fig. 5c). This critical value does not depend on φ2 and φ3 because following treatment, cells T2 and T3 do not acquire new infections and cells I2 and I3 are eventually lost owing to cell death even when IFN fails to cure them. IFN-refractory cells, T1, remain susceptible to new infections even during treatment and thus determine treatment outcome. When their number is sufficiently small, viral production from infected cells is inadequate to replenish the loss of infected cells owing to cell death (akin to the basic reproductive number being below unity50), resulting in the eventual clearance of infection. Thus, the success of IFN treatment depends primarily on the size of the IFN-refractory subpopulation. The larger the latter subpopulation, the smaller the second phase slope, thus presenting an explanation of the strong correlation observed between the second phase slope and treatment response46,51,52.
Comparison with patient data
We next applied our model to analyze data of viral kinetics obtained previously from patients under IFN monotherapy46. Our model provided excellent fits to the data (Fig. 6a, Supplementary Fig. 6, Supplementary Tables 5 and 6). In agreement with our prediction above, we found from the fits that  , an upper bound on φ1 obtained by letting φ2=0, increased from 0.01 for a typical rapid responder, to 0.11 for a slow partial responder and 0.37 for a null responder. Similarly, patients with known favourable haplotypes based on SNPs in the IFN-λ locus had significantly lower
, an upper bound on φ1 obtained by letting φ2=0, increased from 0.01 for a typical rapid responder, to 0.11 for a slow partial responder and 0.37 for a null responder. Similarly, patients with known favourable haplotypes based on SNPs in the IFN-λ locus had significantly lower  on an average than patients with unfavourable haplotypes (Supplementary Fig. 6, Supplementary Table 6). Finally, we estimated
on an average than patients with unfavourable haplotypes (Supplementary Fig. 6, Supplementary Table 6). Finally, we estimated  for a large set of patients under IFN monotherapy51,53 and found it to be negatively correlated with the second phase slope (Fig. 6b), reiterating the dependence of the second phase slope and hence treatment outcome on the frequency of IFN-refractory cells.
for a large set of patients under IFN monotherapy51,53 and found it to be negatively correlated with the second phase slope (Fig. 6b), reiterating the dependence of the second phase slope and hence treatment outcome on the frequency of IFN-refractory cells.

(a) Fits of model predictions of viral load decay following IFN treatment (lines) to previous patient data46 (symbols), where the decay patterns mimic typical null responders (green), slow partial responders (red) and rapid responders (blue). We fixed known model parameter values from the literature46,48,49 (Supplementary Table 4). We let inter-patient variations be determined by the parameters β and V0 and the distribution of cells in the three IFN response phenotypes, φ1, φ2 and φ3. We recognized that φ3 is determined by the viral load decline in the first phase (see text). The available second phase data did not allow the estimation of both φ1 and φ2 uniquely. We therefore assumed φ2=0 and estimated  as the corresponding upper bound on φ1. We thus let β, V0 and
as the corresponding upper bound on φ1. We thus let β, V0 and  be adjustable parameters. The resulting estimates of
be adjustable parameters. The resulting estimates of  , which were robust to variations in the fixed parameters (Supplementary Fig. 11), are mentioned above. The best-fit parameter estimates and measures of goodness of fit are listed in Supplementary Table 5. Other parameter values employed are listed in Supplementary Table 4. (b) Pre-treatment frequency of IFN-refractory cells and the slope of the second phase decline estimated from data in two reports on patients under IFN monotherapy (closed circles: ref. 51; open circles: ref. 53) are strongly correlated (Pearson’s correlation coefficient, R=−0.44, P<0.001). Using the standard model of viral dynamics, the efficacy of IFN, ε, and the death rate of infected cells, δ, were estimated from previously published data of viral kinetics for the patients51,53. The second phase slope is the product δε according to the standard model. Further, ε in the standard model corresponds to φ3 in our model, which yielded
, which were robust to variations in the fixed parameters (Supplementary Fig. 11), are mentioned above. The best-fit parameter estimates and measures of goodness of fit are listed in Supplementary Table 5. Other parameter values employed are listed in Supplementary Table 4. (b) Pre-treatment frequency of IFN-refractory cells and the slope of the second phase decline estimated from data in two reports on patients under IFN monotherapy (closed circles: ref. 51; open circles: ref. 53) are strongly correlated (Pearson’s correlation coefficient, R=−0.44, P<0.001). Using the standard model of viral dynamics, the efficacy of IFN, ε, and the death rate of infected cells, δ, were estimated from previously published data of viral kinetics for the patients51,53. The second phase slope is the product δε according to the standard model. Further, ε in the standard model corresponds to φ3 in our model, which yielded  .
.
Pre- and post-treatment ISG expression and response to IFN
We next examined how treatment response correlated with pre- and post-treatment ISG expression. The pre-treatment frequency of cells refractory to IFN would depend on the level of endogenous IFN production following infection as well as the strength of the effector function of ISGs, or, more generally, the effector function of the IFN network. IFN production stimulates the IFN network, whose effector function determines the propensity to clear HCV. Four possibilities thus arise (Fig. 7).

Pre- and post-treatment steady state positive-strand HCV RNA, ISG mRNA and ISG protein expression levels, in (a), an IFN-sensitive cell with low endogenous IFN level (IFNendo=0.05 U ml−1) and potent IFN effector function (ω=0.5, γ=1), representative of responders, and (b), an IFN-refractory cell with high-endogenous IFN level (IFNendo=100 U ml−1) but poor effector function (ω=20, γ=0.1), representative of nonresponders. The dashed line marks zero. IFITM3 is shown as a representative ISG; other ISGs behave similarly. (c) Schematic of expected responses to IFN-based treatment in patients with different endogenous IFN production levels and potencies of IFN effector function. φ1 is the pre-treatment frequency of IFN-refractory cells,  is the critical pre-treatment frequency of IFN-refractory cells for successful treatment response, ΔV is the fold-decline in viral load and ΔISG is the fold increase in ISG mRNA levels relative to their respective pre-treatment levels. ω and γ represent the fold changes in the repression coefficients of all ISGs and the eIF2B production rate constant, respectively, relative to their baseline values in Supplementary Table 3. Parameters: IFNexo=100 U ml−1. Other parameters are listed in Supplementary Tables 1–3.
is the critical pre-treatment frequency of IFN-refractory cells for successful treatment response, ΔV is the fold-decline in viral load and ΔISG is the fold increase in ISG mRNA levels relative to their respective pre-treatment levels. ω and γ represent the fold changes in the repression coefficients of all ISGs and the eIF2B production rate constant, respectively, relative to their baseline values in Supplementary Table 3. Parameters: IFNexo=100 U ml−1. Other parameters are listed in Supplementary Tables 1–3.
When endogenous IFN production is low (for example, owing to HCV-mediated cleavage of mitochondrial antiviral signaling protein (MAVS)43,54) and IFN effector function potent, infection is established because of inadequate stimulation of the IFN network. ISG levels are predicted to be low in such individuals before treatment (Fig. 7a). Exogenous IFN addition during treatment stimulates the IFN network, which increases ISG expression and, given potent IFN effector function, clears infection. Cells in such individuals predominantly admit Q3 at the level of IFN exposure during treatment. Such individuals are predicted to be rapid responders to IFN treatment (Fig. 7c).
When endogenous IFN production is high but IFN effector function compromised, infection is established despite stimulation of the IFN network. Here ISG levels are high before treatment (Fig. 7b), but HCV continues to thrive because of poor effector function. Cells in such individuals predominantly admit Q1 and addition of exogenous IFN contributes little to the control of infection. Such individuals are predicted to be null responders to treatment (Fig. 7c).
When both endogenous IFN production and ISG effectiveness are low, addition of IFN may increase ISG levels but not necessarily clear HCV given the inadequate effector function. Such individuals appear as partial responders. The fraction of IFN-refractory cells is smaller in such individuals than in null responders, but treatment would fail as long as the fraction is larger than the critical value mentioned above. When both endogenous IFN production and ISG effectiveness are high, infection is likely to be spontaneously cleared; spontaneous clearance has been observed to additionally require strong cytotoxic T-cell responses22, which our study did not consider.
We note that IFN effector function is compromised at steps downstream of the step(s) in the IFN signalling network at which HCV interferes. For instance, HCV interfered above at the level of ISG translation, so that poor effector function arose from low levels of ISG proteins despite high-ISG mRNA levels. In this case, both uninfected and infected cells would display high-pre-treatment ISG mRNA expression in nonresponders. If, instead, HCV were to predominantly inhibit the JAK–STAT pathway, low effector function would arise from low ISG mRNA expression. High-pre-treatment ISG expression in nonresponders would then be restricted to uninfected cells via paracrine signalling.
Synergy between DAAs and IFN
Finally, we examined the origin of the synergy between DAAs and IFN. DAAs such as HCV protease and polymerase inhibitors lower the HCV replication rate and would therefore weaken the control of HCV on the IFN network. To characterize the impact of such a DAA, we identified the steady states of the network at different (dose-dependent) DAA efficacies (Fig. 8a). At low DAA efficacies, the system again exhibits the bistable behaviour observed earlier. As the efficacy increases, the HCV RNA level corresponding to Q1 declines and that corresponding to Q2 increases. Interestingly, beyond a critical efficacy, the HCV-high state is rendered unsustainable and the system jumps to Q3 where HCV is cleared. The DAA thus transforms a bistable cell into a cell that admits Q3 alone. Similarly, a cell that admits Q1 alone could be transformed into one that admits both Q1 and Q3 (Fig. 8b). At the cell population level, this effect of the DAA would manifest as an increase in the fraction of IFN-sensitive cells, φ3, at the expense of IFN-refractory cells, φ1, and thus result in improved treatment response.

(a) Concentration-effect curves showing steady state positive-strand HCV RNA levels for fixed ISG protein levels (blue lines) and ISG protein levels for fixed double-stranded HCV RNA levels (red lines) in the presence of a DAA at different efficacies (εDAA). (b) Steady states of the network as a function of DAA efficacy. In a, ω=1, γ=1. In b, ω=10, γ=0.1. ω and γ represent the fold changes in the repression coefficients of all ISGs and the eIF2B production rate constant, respectively, relative to their baseline values in Supplementary Table 3. Other parameters are listed in Supplementary Tables 1–3.
Discussion
A hallmark of HCV infection has been its persistence in many individuals despite strong ISG expression in the liver and high doses of IFN and ribavirin8,9,10. What underlies this persistence has remained poorly understood. In the present study, we provide a new conceptual basis for understanding HCV persistence and response to IFN-based treatment.
Negative regulators of the IFN system, such as SOCS and USP18, as well as the recently identified IFN-associated immunosuppression have been suggested to contribute to viral persistence despite IFN stimulation6,20,21. While these mechanisms are undoubtedly important, they remain partial determinants of HCV persistence and treatment response because they do not explain when pre-treatment ISG expression would be high. Further, a direct role of HCV in suppressing IFN effector function may be important in determining treatment response because DAAs that suppress HCV replication can induce SVRs in previous null responders to PR. Our study predicts that these additional mechanisms, potentially central to HCV persistence and treatment response, may arise from the systems-level properties of the IFN signalling network modulated by HCV.
Our model of the IFN signalling network predicts that HCV induces bistability in the network. IFN signalling is designed to clear viral infections. HCV interferes with IFN signalling and introduces an alternative steady state where HCV persists. To our knowledge, this is the first instance identified where a virus induces bistability in an innate immune signalling network to achieve persistence. A consequence is that IFN effector function can be compromised despite high-IFN production, allowing viral persistence in the presence of high-ISG expression. Addition of exogenous IFN yields no additional antiviral effect. When more than a critical fraction of cells in an individual is refractory to IFN, treatment would fail. DAAs improve treatment response by suppressing HCV replication, which compromises the ability of HCV to induce bistability in the network, thereby lowering the fraction of cells refractory to IFN.
The IFN signalling network comprises a large number of molecular components that interact via multiple feedback loops5. HCV interferes with the network at several levels6,22. A complex combination of molecular players as well as parameter values determining the strengths of their interactions may thus underlie the phenotypic response of an HCV-infected cell to IFN. Currently, the limited knowledge of cell-to-cell and inter-patient variations in parameter values (for example, see ref. 55) precludes a priori estimation of the prevalence of IFN-refractory cells in patients and hence the establishment of a quantitative link between network properties and viral kinetics. Nonetheless, we found that HCV-induced bistability is a robust emergent property of the network: in addition to the translational block induced by HCV considered above, with which current in vivo data29 seems consistent, we examined other known modes of HCV interference of the IFN network and also negative regulation of IFN stimulation by ISG proteins. We found bistability in all cases (Supplementary Notes 7 and 8, Supplementary Figs 7 and 8). Bistability was also maintained under parameter regimes that mimicked the dynamic oscillations in HCV-induced translational inhibition observed recently56,57 (Supplementary Note 9, Supplementary Fig. 9). Further, our prediction that the pre-treatment prevalence of IFN-refractory cells beyond a critical fraction leads to treatment failure is independent of the origin of the bistability.
Previous models of viral kinetics have estimated the effectiveness of PR by comparisons of model predictions with viral load decline in patients14,46,47,48,49,58. Our model reproduced all the patterns of viral kinetics observed in patients by varying the fractions of cells refractory and sensitive to IFN. Alternative interpretations thus emerge of the effectiveness estimated by previous models. The extent of first phase decline, which is taken as a measure of IFN effectiveness in lowering viral production, is in our model a measure of the pre-treatment fraction of infected cells that are subsequently cured by PR. The slope of the second phase, which is taken as a combination of PR effectiveness and the loss rate of infected cells, is in our model a combination of the loss rate of infected cells and the fraction of cells refractory to PR. Larger the latter fraction, smaller the slope, and hence poorer the treatment response. The critical effectiveness of PR predicted by previous models47 for successful treatment translates to the level of PR activity necessary to render the fraction of cells refractory to PR below the critical value identified by our model. Our model thus presents an alternative yet equally consistent description of viral kinetics and one that is rooted in the properties of the IFN network.
The synergy between DAAs and IFN predicted by our model can be compromised by the emergence of resistance to DAAs, which we did not consider. Recent studies have reported the dependence of viral kinetics on SNPs even in IFN-free treatment regimens59,60 and recent trials with some two-DAA combinations without PR have been stunningly successful with >90% SVR in previous null responders to PR61. We speculate that this success may arise from a combination of three modes of DAA action. First, the DAAs may directly suppress HCV replication. Second, they may render cells monostable and facilitate HCV clearance. Third, the resulting lower HCV levels may lead to lower IFN stimulation, which in the absence of exogenous IFN may undo the immunosuppression caused by persistent IFN signalling and effect a cure. Identifying which of these mechanisms predominates in vivo awaits new investigations and may inform future strategies of intervention.
Methods
Mathematical model of the IFN signalling network
We modelled the network (Fig. 1) based on mass action and Michaelis–Menten kinetics (Supplementary Note 1). The parameters involved (Supplementary Tables 1–3) were either obtained from the literature or estimated from analysis of independent experiments of intracellular HCV RNA kinetics (Supplementary Figs 3 and 10), ISG expression following stimulation with IFN, and suppression of HCV RNA by over-expression of ISGs (Supplementary Note 2). The fit of this model to the highly infectious HCV JFH-m4 strain can be found in Supplementary Fig. 10 and Supplementary Note 2.
Mathematical model of viral kinetics
We modified the standard model of HCV viral kinetics to incorporate subpopulations of cells with distinct phenotypic responses to IFN (Fig. 4, Supplementary Note 5). We obtained analytical approximations to the resulting first and early second phase slopes (Supplementary Note 6). The parameter values employed were obtained from the literature or estimated from fits to patient data (Supplementary Tables 4–6).
Solution of model equations and data analysis
We solved the model of the IFN signalling network and the model of viral kinetics using the Rosenbrock algorithm in Berkeley-Madonna version 8.3.18 and the ode15s routine in MATLAB. We fit the model predictions to the data using the nonlinear regression tool NLINFIT in MATLAB and estimated 95% confidence intervals using the tool NLPARCI in MATLAB. We assessed the goodness of fits using R2 and the Student’s t-test on residuals. All data digitization was done using Engauge digitizer. The computer programs employed are available upon request.
Additional information
How to cite this article: Padmanabhan, P. et al. Emergent properties of the interferon-signalling network may underlie the success of hepatitis C treatment. Nat. Commun. 5:3872 doi: 10.1038/ncomms4872 (2014).
References
Lavanchy, D. Evolving epidemiology of hepatitis C virus. Clin. Microbiol. Infect. 17, 107–115 (2011).
Ghany, M. G., Nelson, D. R., Strader, D. B., Thomas, D. L. & Seeff, L. B. An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 54, 1433–1444 (2011).
Kau, A., Vermehren, J. & Sarrazin, C. Treatment predictors of a sustained virologic response in hepatitis B and C. J. Hepatol. 49, 634–651 (2008).
Hayes, C. N., Imamura, M., Aikata, H. & Chayama, K. Genetics of IL28B and HCV-response to infection and treatment. Nat. Rev. Gastroenterol. Hepatol. 9, 406–417 (2012).
Haller, O., Kochs, G. & Weber, F. The interferon response circuit: induction and suppression by pathogenic viruses. Virology 344, 119–130 (2006).
Heim, M. H. Innate immunity and HCV. J. Hepatol. 58, 564–574 (2013).
Micallef, J. M., Kaldor, J. M. & Dore, G. J. Spontaneous viral clearance following acute hepatitis C infection: a systematic review of longitudinal studies. J. Viral Hepat. 13, 34–41 (2006).
Lau, D. T. et al. Innate immune tolerance and the role of Kupffer cells in differential responses to interferon therapy among patients with HCV genotype 1 infection. Gastroenterology 144, 402–413 (2013).
Sarasin-Filipowicz, M. et al. Interferon signaling and treatment outcome in chronic hepatitis C. Proc. Natl Acad. Sci. USA 105, 7034–7039 (2008).
Feld, J. J. et al. Hepatic gene expression during treatment with peginterferon and ribavirin: identifying molecular pathways for treatment response. Hepatology 46, 1548–1563 (2007).
Di Bisceglie, A. M. et al. Ribavirin as therapy for chronic hepatitis C. A randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 123, 897–903 (1995).
Fried, M. W. et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 347, 975–982 (2002).
Rotman, Y. et al. Effect of ribavirin on viral kinetics and liver gene expression in chronic hepatitis C. Gut 63, 161–169 (2013).
Dixit, N. M., Layden-Almer, J. E., Layden, T. J. & Perelson, A. S. Modelling how ribavirin improves interferon response rates in hepatitis C virus infection. Nature 432, 922–924 (2004).
Thomas, E. et al. Ribavirin potentiates interferon action by augmenting interferon-stimulated gene induction in hepatitis C virus cell culture models. Hepatology 53, 32–41 (2011).
Welsch, C., Jesudian, A., Zeuzem, S. & Jacobson, I. New direct-acting antiviral agents for the treatment of hepatitis C virus infection and perspectives. Gut 61, (Suppl 1): i36–i46 (2012).
Pawlotsky, J. M. Treatment failure and resistance with direct-acting antiviral drugs against hepatitis C virus. Hepatology 53, 1742–1751 (2011).
Muir, A. J. et al. Retreatment with telaprevir combination therapy in hepatitis C patients with well-characterized prior treatment response. Hepatology 54, 1538–1546 (2011).
McHutchison, J. G. et al. Telaprevir for previously treated chronic HCV infection. N. Engl. J. Med. 362, 1292–1303 (2010).
Teijaro, J. R. et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340, 207–211 (2013).
Wilson, E. B. et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340, 202–207 (2013).
Thimme, R., Binder, M. & Bartenschlager, R. Failure of innate and adaptive immune responses in controlling hepatitis C virus infection. FEMS Microbiol. Rev. 36, 663–683 (2012).
Schoggins, J. W. et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485 (2011).
Metz, P. et al. Identification of type I and type II interferon-induced effectors controlling hepatitis C virus replication. Hepatology 56, 2082–2093 (2012).
Raychoudhuri, A. et al. ISG56 and IFITM1 proteins inhibit hepatitis C virus replication. J. Virol. 85, 12881–12889 (2011).
Kim, M. J. & Yoo, J. Y. Inhibition of hepatitis C virus replication by IFN-mediated ISGylation of HCV-NS5A. J. Immunol. 185, 4311–4318 (2010).
Han, J. Q. & Barton, D. J. Activation and evasion of the antiviral 2′-5′ oligoadenylate synthetase/ribonuclease L pathway by hepatitis C virus mRNA. RNA 8, 512–525 (2002).
Garaigorta, U. & Chisari, F. V. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe 6, 513–522 (2009).
Wieland, S. et al. Simultaneous detection of hepatitis C virus and interferon stimulated gene expression in infected human liver. Hepatology http://dx.doi.org/10.1002/hep.26770 (2013).
Perales, C. et al. Response of hepatitis C virus to long-term passage in the presence of interferon-alpha. Multiple mutations and a common phenotype. J. Virol. 87, 7593–7607 (2013).
Proud, C. G. eIF2 and the control of cell physiology. Semin. Cell Dev. Biol. 16, 3–12 (2005).
Rowlands, A. G., Panniers, R. & Henshaw, E. C. The catalytic mechanism of guanine nucleotide exchange factor action and competitive inhibition by phosphorylated eukaryotic initiation factor 2. J. Biol. Chem. 263, 5526–5533 (1988).
Dabo, S. & Meurs, E. F. dsRNA-dependent protein kinase PKR and its role in stress, signaling and HCV infection. Viruses 4, 2598–2635 (2012).
Cole, J. L. Activation of PKR: an open and shut case? Trends Biochem. Sci. 32, 57–62 (2007).
Koev, G., Duncan, R. F. & Lai, M. M. Hepatitis C virus IRES-dependent translation is insensitive to an eIF2alpha-independent mechanism of inhibition by interferon in hepatocyte cell lines. Virology 297, 195–202 (2002).
Shimoike, T., McKenna, S. A., Lindhout, D. A. & Puglisi, J. D. Translational insensitivity to potent activation of PKR by HCV IRES RNA. Antivir. Res. 83, 228–237 (2009).
Terenin, I. M., Dmitriev, S. E., Andreev, D. E. & Shatsky, I. N. Eukaryotic translation initiation machinery can operate in a bacterial-like mode without eIF2. Nat. Struct. Mol. Biol. 15, 836–841 (2008).
Ferrell, J. E. & Xiong, W. Bistability in cell signaling: how to make continuous processes discontinuous, and reversible processes irreversible. Chaos 11, 227–236 (2001).
Das, J. et al. Digital signaling and hysteresis characterize Ras activation in lymphoid cells. Cell 136, 337–351 (2009).
Bhalla, U. S. & Iyengar, R. Emergent properties of networks of biological signaling pathways. Science 283, 381–387 (1999).
Acar, M., Becskei, A. & van Oudenaarden, A. Enhancement of cellular memory by reducing stochastic transitions. Nature 435, 228–232 (2005).
Alon, U. Network motifs: theory and experimental approaches. Nat. Rev. Genet. 8, 450–461 (2007).
Cheng, G., Zhong, J. & Chisari, F. V. Inhibition of dsRNA-induced signaling in hepatitis C virus-infected cells by NS3 protease-dependent and -independent mechanisms. Proc. Natl Acad. Sci. USA 103, 8499–8504 (2006).
Rand, U. et al. Multi-layered stochasticity and paracrine signal propagation shape the type-I interferon response. Mol. Syst. Biol. 8, 584 (2012).
Levin, D., Harari, D. & Schreiber, G. Stochastic receptor expression determines cell fate upon interferon treatment. Mol. Cell Biol. 31, 3252–3266 (2011).
Neumann, A. U. et al. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science 282, 103–107 (1998).
Dahari, H., Ribeiro, R. M. & Perelson, A. S. Triphasic decline of hepatitis C virus RNA during antiviral therapy. Hepatology 46, 16–21 (2007).
Rong, L., Dahari, H., Ribeiro, R. M. & Perelson, A. S. Rapid emergence of protease inhibitor resistance in hepatitis C virus. Sci. Transl. Med. 2, 30ra32 (2010).
Guedj, J. & Perelson, A. S. Second-phase hepatitis C virus RNA decline during telaprevir-based therapy increases with drug effectiveness: implications for treatment duration. Hepatology 53, 1801–1808 (2011).
Nowak, M. A. & May, R. M. inVirus Dynamics: Mathematical Principles of Immunology and Virology Oxford Univ. Press (2000).
Layden, J. E. et al. First phase viral kinetic parameters as predictors of treatment response and their influence on the second phase viral decline. J. Viral Hepat. 9, 340–345 (2002).
Mihm, U., Herrmann, E., Sarrazin, C. & Zeuzem, S. Predicting response in hepatitis C virus therapy. Aliment. Pharmacol. Ther. 23, 1043–1054 (2006).
Ribeiro, R. M., Layden-Almer, J., Powers, K. A., Layden, T. J. & Perelson, A. S. Dynamics of alanine aminotransferase during hepatitis C virus treatment. Hepatology 38, 509–517 (2003).
Bellecave, P. et al. Cleavage of mitochondrial antiviral signaling protein in the liver of patients with chronic hepatitis C correlates with a reduced activation of the endogenous interferon system. Hepatology 51, 1127–1136 (2010).
Ireton, R. C. & Gale, M. Jr Systems biology analyses to define host responses to HCV infection and therapy. Curr. Top. Microbiol. Immunol. 363, 143–167 (2013).
Garaigorta, U., Heim, M. H., Boyd, B., Wieland, S. & Chisari, F. V. Hepatitis C virus (HCV) induces formation of stress granules whose proteins regulate HCV RNA replication and virus assembly and egress. J. Virol. 86, 11043–11056 (2012).
Ruggieri, A. et al. Dynamic oscillation of translation and stress granule formation mark the cellular response to virus infection. Cell. Host. Microbe. 12, 71–85 (2012).
Guedj, J. et al. Modelling shows that the NS5A inhibitor daclatasvir has two modes of action and yields a shorter estimate of the hepatitis C virus half-life. Proc. Natl Acad. Sci. USA 110, 3991–3996 (2013).
Chu, T. W. et al. Effect of IL28B genotype on early viral kinetics during interferon-free treatment of patients with chronic hepatitis C. Gastroenterology 142, 790–795 (2012).
Meissner, E. G. et al. IFNL4-deltaG genotype is associated with slower viral clearance in hepatitis C, genotype-1 patients treated with sofosbuvir and ribavirin. J. Infect. Dis. http://dx.doi.org/10.1093/infdis/jit1827 (2014).
Hagan, L. M. & Schinazi, R. F. Best strategies for global HCV eradication. Liver Int. 33, 68–79 (2013).
Acknowledgements
This work was supported by the Department of Science and Technology, Government of India. We thank Rahul Roy for comments.
Author information
Authors and Affiliations
Contributions
P.P. and N.M.D. conceived the study and wrote the manuscript. P.P. performed the study. P.P., U.G. and N.M.D. analyzed the data, and contributed tools and reagents.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-11, Supplementary Tables 1-6, Supplementary Notes 1-9 and Supplementary References (PDF 1811 kb)
Rights and permissions
About this article

Cite this article
Padmanabhan, P., Garaigorta, U. & Dixit, N. Emergent properties of the interferon-signalling network may underlie the success of hepatitis C treatment. Nat Commun 5, 3872 (2014). https://doi.org/10.1038/ncomms4872
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms4872
This article is cited by
-
Modeling how antibody responses may determine the efficacy of COVID-19 vaccines
Nature Computational Science (2022)
-
Super-resolution microscopy: a closer look at synaptic dysfunction in Alzheimer disease
Nature Reviews Neuroscience (2021)
-
Direct-acting antiviral treatment downregulates immune checkpoint inhibitor expression in patients with chronic hepatitis C
Clinical and Experimental Medicine (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
