
Abstract
Myosin 1b is a single-headed membrane-associated motor that binds to actin filaments with a catch-bond behaviour in response to load. In vivo, myosin 1b is required to form membrane tubules at both endosomes and the trans-Golgi network. To establish the link between these two fundamental properties, here we investigate the capacity of myosin 1b to extract membrane tubes along bundled actin filaments in a minimal reconstituted system. We show that single-headed non-processive myosin 1b can extract membrane tubes at a biologically relevant low density. In contrast to kinesins we do not observe motor accumulation at the tip, suggesting that the underlying mechanism for tube formation is different. In our theoretical model, myosin 1b catch-bond properties facilitate tube extraction under conditions of increasing membrane tension by reducing the density of myo1b required to pull tubes.
Similar content being viewed by others


Introduction
Understanding the mechanisms that cells use to produce physical forces to control their own shape and the morphology of their compartments is a fundamental problem of cell biology. Because of their ability to pull on membranes and transport vesicles or organelles1,2, microtubule-associated motor proteins are key factors in membrane remodelling3. Indeed, in vitro, these motors can pull membrane tubular networks along microtubules from Golgi or endoplasmic reticulum membranes4,5,6. In addition, processive motors, that is, motors making multiple consecutive steps on their cytoskeleton substrate, such as kinesin-1, are able to cooperatively extract nanotubes along microtubules by exerting a force on the membrane to which they are bound7,8,9. Growing evidence indicates that actin-based motors, such as myosins 5b, 6 and 1b, are also involved in membrane tubule formation in vivo10,11,12,13. We previously reported that myosin 1b (myo1b), although non-processive, that is, it detaches from actin filament during each ATPase cycle and makes a single step only, is indispensable for the formation of post-Golgi tubular carriers from the trans-Golgi network (TGN) and modulates the membrane tube extension from sorting endosomes12,13. Similar to the other members of the myosin 1 family, myo1b is a membrane-associated single-headed motor that binds membrane via its carboxy terminal domain also named tail. This region presents a highly basic tail homology 1 (TH1) domain that targets myosins 1 to lipid membranes and a pleckstrin homology motif in the TH1 domain that targets myo1b to the membrane containing phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2)14,15. Myosins 1 are non-processive but some members of this family, including myo1b, have a catch-bond behaviour (a decreasing detachment rate on increasing load)16,17,18 in contrast, for example, to kinesin-1, which has a slip-bond behaviour (an increasing detachment rate with increasing load)19. Despite the crucial role of myo1b in tubule formation established in vivo, the understanding of the mechanism by which a single-headed, non-processive and force-sensitive motor contributes to the tubule formation is lacking. Two fundamental questions can be raised: (i) is myo1b able and sufficient to pull membrane tubes along actin filaments? (ii) more generally, how can a non-processive motor elongates a membrane tube? Here we show using a reconstituted system that myo1b can extract tubes from giant unilamellar vesicle (GUV) containing PI(4,5)P2. We propose a theoretical model that emphasizes the importance of myo1b catch-bond properties for facilitation of tube extraction.
Results
Myo1b elongates tubes from GUVs
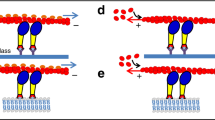
We first reconstituted a minimal system composed of purified full-length recombinant myo1b, GUV containing PI(4,5)P2 as a model membrane and actin filaments (F-actin) bundled with a uniform polarity on a glass substrate as tracks for myo1b (Fig. 1). Instead of artificially binding molecular motors to GUVs via biotin–streptavidin coupling as it was previously done with kinesins or NCD, we took advantage of myo1b biologically relevant interactions with PI(4,5)P2 lipids and bound the full-length motor with GUVs via these lipids.

Membrane tubes grow from a giant unilamellar vesicle (GUV) coated with myosins 1b (myo1b) sedimented on actin filaments bundled by fascin immobilized on a glass substrate.
We produced and purified recombinant myo1b using the Flp-In system13 that allows to obtain proteins on a large scale from mammalian cell culture (Methods). Analysis by gel electrophoresis of purified myo1b demonstrated a strong enrichment in myo1b (Supplementary Fig. 1). The analysis by cryo-EM of the preparation showed isolated molecules of 27 nm length on average as previously reported for myo1b20 (Fig. 2a). We also found myo1b bound to rare native vesicles present in the preparation (Fig. 2b), demonstrating the capacity of myo1b to bind membranes in vivo. Moreover, the motor property of recombinant myo1b was preserved after purification and fluorescent labelling, as judged by F-actin gliding motility assays. Fluorescent myo1b was able to move fluorescent F-actin at room temperature (RT) and in the presence of 2 mM ATP with an average velocity of 56.0±4.3 nm s−1 (Supplementary Fig. 2), in the same range as those previously reported for myo1b21. We next investigated the ability of myo1b to bind small liposomes in a fluid phase, generated from a lipid film composed of egg yolk L-α-phosphatidylcholine (egg PC), cholesterol and in the presence or the absence of PI(4,5)P2. Analysis by cryo-EM revealed that myo1b bound to the liposomes containing PI(4,5)P2 (Fig. 2c and Supplementary Fig. 3A versus B). Although myo1b molecules were densely distributed, forming a brush-like structure, no clustering of the motors was observed at the surface of the liposomes (Fig. 2c; Supplementary Fig. 3B and C). Overall, these data showed that purified myo1b was functional and able to interact with the PI(4,5)P2-containing membrane.

(a) Cryo-EM image of myo1b (white arrows pointing to the head groups). The dark network is the carbon of the holey EM grid. (b) Cryo-EM image of one of the rare native vesicles copurified with myo1b. Myosin heads point 27 nm outwards the lipid bilayer. (Inset) Immunolabelling of myo1b bound to native vesicles. (c) Cryo-EM image of myo1b bound on a small synthetic liposome made of egg PC/cholesterol/PI(4,5)P2 incubated with purified recombinant myo1b. Despite the high protein density, no myosin cluster is observed. (d,e) Confocal microscopy image of myo1b bound to a PI(4,5)P2-containing GUV. Labelling corresponds to (d) 0.3% Texas Red DHPE (mol/mol) and (e) Alexa Fluor 488 conjugated myo1b. Scale bars, (a–c) 50 nm, (d,e) 10 μm. White arrows: equatorially bound myosins. White star: carbon network of the holey grids.
We next analysed the formation of tubes on GUVs by optical microscopy. Two hundred nanomolars of myo1b was then incubated with GUVs, generated by electroformation from lipid films with the same composition as that of the small liposomes, supplemented with 0.3% Texas Red 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (Texas Red DHPE) (mol/mol). In all, 10–20% of the GUVs showed a homogeneous fluorescence that does not result from the interaction of free fluorescent dye with the membrane (Supplementary Fig. 4) but was due to myo1b binding (Fig. 2d,e). Furthermore, no binding was found in the absence of PI(4,5)P2 (Supplementary Figs 3A and 5). We prepared F-actin bundled with fascin, leading to polar bundles. After sedimentation of GUVs covered with myo1b on F-actin bundles randomly fixed on a poly-L-lysine-coated glass slide, we monitored tube extension at the bottom of the GUVs in contact with the actin bundles using confocal microscopy. We observed membrane tube growth along F-actin bundles in the presence of ATP (Fig. 3a and Supplementary Movie 1). Myo1b, membrane and F-actin were co-localized as shown by fluorescence microscopy (Fig. 3b). Tubes do not grow by passive spreading of the membrane decorated with myo1b on F-actin since replacement of 2 mM ATP by 2 mM ADP inhibited extraction (Supplementary Fig. 6). Myo1b ATPase activity and its catch-bond property are regulated by calcium concentration, as calcium releases calmodulin—a regulatory myosin light chain—from myo1b21,22. In agreement with these reports, we observed that complementing our assay with 0.5 mM calcium (in the absence of EGTA) totally inhibited tube extraction (Fig. 4a). Furthermore, addition of 2 mM calcium (in the presence of 1 mM EGTA) during tube extension induced fast retraction (Fig. 4b). The effect was not due to an osmotic shock. The osmolarity of the calcium solution was carefully adjusted to the same value as the assay buffer. Moreover, the distribution of fluorescent myo1b remained homogeneous on calcium addition (Supplementary Fig. 7), although calcium is known to cluster PI(4,5)P2 in certain conditions23,24. This demonstrates that active myo1b are responsible for tube growth.

(a) Growth of membrane tubes between a reference time t (top) and t+5 min (bottom) followed with confocal microscopy. Confocal sections at the bottom of the GUV where the GUV is in contact with the substrate covered with actin bundles are shown. Zoom of the box is shown in d. (b) Images of the same tube at t+11 min showing the distribution of lipid (magenta), myo1b (green) and actin (blue) (c) Growth kinetics of membrane tubes as a function of myo1b total density (actin bundle-bound and - unbound motors). Motor density is represented according to the colour scale represented on the right. Data at low density are binned over three points. (d) Zoom of the tip of the tube (box in a) showing confocal fluorescence microscopy images (left) and intensity profiles (bottom right) of lipid membrane and myo1b. Scale bars, 5 μm.

(a) Calcium prevents myo1b from extracting tubes: confocal microscopy images of lipid (magenta), myo1b (green) and actin (blue) at the bottom of a GUV in the presence of 0.5 mM calcium. No tube extraction was observed. (b) Addition of calcium induces retraction from a growing tube: confocal microscopy images of lipid (magenta), myo1b (green) and actin (blue) during tube growth (left) and 20 s after the addition of 2 mM calcium (right). Note that tube retraction is much faster than tube extension. Scale bar, 5 μm.
Tube elongation velocity and myo1b density along the tubes were determined using time-lapse confocal microscopy (Methods). Myo1b density on the membrane has been measured from fluorescence analysis on the tubes (Methods). Remarkably, although myo1b is non-processive, directional tube growth was observed with myo1b densities of the same order of magnitude as the threshold density for processive (kinesin-1) or non-processive (NCD) kinesins (as low as 300 motors μm−2)9,25,26. Myo1b-associated tubes grew regularly with an average velocity ranging from 5 to 100 nm s−1 (typical elongation plots are shown in Fig. 3c) with myo1b density ranging from 300 motors μm−2 to 5,200 motors μm−2 (see below and Supplementary Movies 1–3). Furthermore, Myo1b did not accumulate at the tube tip (Fig. 3d) in contrast to kinesins pulling tubes9,25. Finally, in contrast with NCD25, we never observed bidirectional growth of the tubes. This suggests different underlying mechanisms of tube formation for single-headed non-processive myosins and processive or non-processive kinesins, and thus requires the development of a new theoretical model to understand this process.
Myo1b catch-bond behaviour facilitates tubule elongation
Our theoretical model makes the following hypotheses (see Fig. 5a for a sketch): (i) myo1b remains attached to the membrane due to its high affinity Kd=6 nM (ref. 15); its detachment rate is insensitive to load in the range of forces investigated and thus neglected; myo1b diffuses along the tube surface and binds transiently to F-actin during the ATPase cycle. Thus, binding/unbinding refers only to myo1b and F-actin, but not to myo1b and membrane. (ii) As the membrane is fluid, myo1b exerts a force only at the tube tip9. (iii) Being non-processive, myo1b performs only one step after binding to F-actin; membrane tube extension requires, therefore, the binding of another motor ahead of the leading motor; yet myo1b being a catch-bond motor, its binding time increases with increasing load. Thus, tube dynamics results from binding and unbinding kinetics of the tip motor. The growth is facilitated by an increase in myo1b density and by applying a load on the myo1b. The unbinding rate of the catch-bond myo1b from actin filaments ωoff decreases exponentially with load f ωoff(f)=1.6exp(−f.ddet/kBT)+0.02 s−1 with, kBT/ddet=0.3 pN17 (Fig. 5a) (see details on the microscopic model for myo1b ATPase cycle in Supplementary Note). In the following, we use ωoff as ωoff(0). We call nf the number of filaments in the actin bundle accessible to the myo1b motors, which corresponds to a fraction of the total number. Motor binding on F-actin is possible if one of the nf filaments of the actin bundle is not already occupied. We write the binding rate as  where d≈5 nm is the motor step, R0 is the membrane tube radius and ρb the surface density of bound motors. We suppose here that the binding rate of an isolated motor on a single filament ωon (Fig. 5a) is independent of applied load f. The unbound motors of surface density ρu diffuse along the tube with a diffusion constant D (Fig. 5a), whereas the bound motors of density ρb perform their powerstroke before unbinding. The kinetic equations for a tube advancing at velocity
where d≈5 nm is the motor step, R0 is the membrane tube radius and ρb the surface density of bound motors. We suppose here that the binding rate of an isolated motor on a single filament ωon (Fig. 5a) is independent of applied load f. The unbound motors of surface density ρu diffuse along the tube with a diffusion constant D (Fig. 5a), whereas the bound motors of density ρb perform their powerstroke before unbinding. The kinetic equations for a tube advancing at velocity  are:
are:

(a) Sketch of the model with D the diffusion coefficient of the motors (green) on the membrane (magenta),  the tube growth velocity, ωon/ωoff the binding/unbinding rates of motors on the actin bundle and R0 the tube radius. The motors bound at the tip support the load on the tube. The tube length L increases by one step (blue dashed line) if a motor binds (blue arrow) further on the actin bundle (blue). If the last motor bound at the tip unbinds (red arrow), the tube retracts until it reaches a bound motor (red dashed line). (b) Phase diagram of tube extraction as a function of membrane tension σ (or equivalently the plateau force f0, as σ=1/2κ(f0/2π)2) and the motor density on the vesicle ρv rescaled with the number of F-actin in the bundles nf (see Supplementary Note) (n=11). In the region 1, no tube is pulled (circle). In 2, tubes are systematically extracted (circle+tube). In 3, tubes are extracted stochastically. 3 is subdivided in two parts, corresponding to extraction time larger or lower than 100 s. Experimentally observed tubes (black crosses) are in 2 or in the ‘fast’ region 3. (c) Growth velocity
the tube growth velocity, ωon/ωoff the binding/unbinding rates of motors on the actin bundle and R0 the tube radius. The motors bound at the tip support the load on the tube. The tube length L increases by one step (blue dashed line) if a motor binds (blue arrow) further on the actin bundle (blue). If the last motor bound at the tip unbinds (red arrow), the tube retracts until it reaches a bound motor (red dashed line). (b) Phase diagram of tube extraction as a function of membrane tension σ (or equivalently the plateau force f0, as σ=1/2κ(f0/2π)2) and the motor density on the vesicle ρv rescaled with the number of F-actin in the bundles nf (see Supplementary Note) (n=11). In the region 1, no tube is pulled (circle). In 2, tubes are systematically extracted (circle+tube). In 3, tubes are extracted stochastically. 3 is subdivided in two parts, corresponding to extraction time larger or lower than 100 s. Experimentally observed tubes (black crosses) are in 2 or in the ‘fast’ region 3. (c) Growth velocity  versus ρv (deduced from Supplementary equation (4)) from experiments (n=12). Error bars represent SD × 0.67, corresponding to 50% confidence interval (see Supplementary for details on error bars). The experimental points are bound by the theoretical curves (equation (2)) (red) with the catch-bond activated, ρv=ρu and nf =1 and 6. (d) Theoretical variation of
versus ρv (deduced from Supplementary equation (4)) from experiments (n=12). Error bars represent SD × 0.67, corresponding to 50% confidence interval (see Supplementary for details on error bars). The experimental points are bound by the theoretical curves (equation (2)) (red) with the catch-bond activated, ρv=ρu and nf =1 and 6. (d) Theoretical variation of  versus ρv (Eq. (2)) for three different forces f=0 (blue), 0.3 (red) and 1 pN (black), and nf=4. The arrows correspond to the respective motor density thresholds
versus ρv (Eq. (2)) for three different forces f=0 (blue), 0.3 (red) and 1 pN (black), and nf=4. The arrows correspond to the respective motor density thresholds  .
.

In our experiments, we only measure the total density ρ=ρb+ρu. Our model predicts that ρ is constant if  (with r=ρb/ρ) and t* larger than 103 s in our experimental conditions (Supplementary Note). Therefore, the surface density on the vesicle ρv is equal to the density along the tube, ρv=ρu (Supplementary Note). Moreover, in agreement with our observations no extended motor accumulation at the tip as observed for tubes pulled by kinesins is expected.
(with r=ρb/ρ) and t* larger than 103 s in our experimental conditions (Supplementary Note). Therefore, the surface density on the vesicle ρv is equal to the density along the tube, ρv=ρu (Supplementary Note). Moreover, in agreement with our observations no extended motor accumulation at the tip as observed for tubes pulled by kinesins is expected.
In the Supplementary Note, we model the elongation of the tube as a Markov process with advancing and receding steps. The elongation velocity reads:

where P(1) is the probability to find a single motor at the tip. The first term is the advancing velocity due to the binding of new motors elongating the tube by d, whereas the second term is the retraction velocity due to the unbinding of the first motor (Supplementary Note). The retraction step is d/1−q where  because the tube retracts until another bound motor sustains the force. The elongation velocity decreases with decreasing motor concentration and vanishes at
because the tube retracts until another bound motor sustains the force. The elongation velocity decreases with decreasing motor concentration and vanishes at  . Because of the catch-bond effect, the critical density decreases with the membrane force, by a factor 10 for a few pN force.
. Because of the catch-bond effect, the critical density decreases with the membrane force, by a factor 10 for a few pN force.
We next determined the conditions for tube extraction that we summarized in the phase diagram of Fig. 5b. The force required to pull a tube from a vesicle increases linearly with the amplitude of the vesicle deformation and reaches a plateau when the tube is formed27. The plateau force f0 and the radius R0 depend on the membrane bending modulus κ and the vesicle tension σ, f0=2π(2σκ)1/2 and R0=(κ/(2σ))1/2 (ref. 27). If the motor density on the vesicle is larger than the critical density at vanishing force  , myo1b systematically extracts a tube (Fig. 5b, region 2). If the motor concentration is such that
, myo1b systematically extracts a tube (Fig. 5b, region 2). If the motor concentration is such that  , tubes do not have a positive velocity at vanishing force. Thus, no tube extraction occurs until fluctuations of motors binding to F-actin produce a membrane deformation sufficient to initiate tube elongation. By analogy with a particle in a metastable energy well, we describe this region of the phase diagram as stochastic extraction (Fig. 5b, region 3). In this regime, we calculated the average time for tube extraction as a first passage problem (Supplementary Note). Finally, if
, tubes do not have a positive velocity at vanishing force. Thus, no tube extraction occurs until fluctuations of motors binding to F-actin produce a membrane deformation sufficient to initiate tube elongation. By analogy with a particle in a metastable energy well, we describe this region of the phase diagram as stochastic extraction (Fig. 5b, region 3). In this regime, we calculated the average time for tube extraction as a first passage problem (Supplementary Note). Finally, if  , no tube is extracted (Fig. 5b, region 1). In region 3 characteristic of catch-bond, we distinguish two subregions depending on whether the average extraction time is larger or smaller than the experimental time 100 s (region 3, Fig. 5b). We consider that the probability of extraction vanishes if the extraction time is larger than 100 s.
, no tube is extracted (Fig. 5b, region 1). In region 3 characteristic of catch-bond, we distinguish two subregions depending on whether the average extraction time is larger or smaller than the experimental time 100 s (region 3, Fig. 5b). We consider that the probability of extraction vanishes if the extraction time is larger than 100 s.
The experimental conditions under which tubes are extracted are well supported by the calculated phase diagram (Fig. 5b). As demonstrated in ref. 27, the force to pull a tube can be calculated from the membrane bending rigidity and tube radius, regardless of the vesicle morphology, as the theory was developed for a patch of membrane. Consequently, both the plateau force and the membrane tension for each tube were calculated from  and
and  where the radius R0 was deduced from fluorescence analysis (Methods) and κ from independent measurements κ=31.7±3.0 kBT=127±12 pN nm (Methods and Supplementary Fig. 8). As the unbinding rate is expected to increase when exceeding a force fm (ref. 28), in agreement with experiments, we set fm=35 pN. No tube can be extracted under these conditions (Fig. 5b, region 1). All vesicles generating tubes in our experiments correspond to region 2 of spontaneous tube growth of the phase diagram or to subregion 3 (average extraction time smaller than 100 s). In region 2, several tubes can be extracted from the same GUV, whereas in region 3 only one tube is extracted (Fig. 5b and Supplementary Movie 2).
where the radius R0 was deduced from fluorescence analysis (Methods) and κ from independent measurements κ=31.7±3.0 kBT=127±12 pN nm (Methods and Supplementary Fig. 8). As the unbinding rate is expected to increase when exceeding a force fm (ref. 28), in agreement with experiments, we set fm=35 pN. No tube can be extracted under these conditions (Fig. 5b, region 1). All vesicles generating tubes in our experiments correspond to region 2 of spontaneous tube growth of the phase diagram or to subregion 3 (average extraction time smaller than 100 s). In region 2, several tubes can be extracted from the same GUV, whereas in region 3 only one tube is extracted (Fig. 5b and Supplementary Movie 2).
We then compared theory and experimental data for the growth velocity as a function of the motor density on the vesicle ρv. Experimentally, we can measure the protein density on the tube ρ and the tube radius R0; the density ρv is calculated by solving Supplementary equation (4) (see Supplementary Note). As predicted by using equation (2), the experimental mean velocity increases with density (Fig. 5c) and even exceeds the velocity of a free F-actin gliding on myo1b (See Supplementary Note for more details). The number of actin filaments in the bundles involved in tube growth cannot be directly measured. With ωon=1.5 × 10−3 μm2 s−1, we found that nf ranges between 1 and 6 in our samples (Fig. 5c), a narrow range in agreement with ref. 29, which is consistent with our assumption that ωon is independent of applied force. The binding rate for a membrane saturated with motors is on the order of a few s−1, in agreement with the phosphate release rate measured in ref. 17. We plotted the theoretical variation of the tube velocity  versus ρv to evaluate the effect of catch-bond on the critical density
versus ρv to evaluate the effect of catch-bond on the critical density  . On the basis of our experiments, we used the median value nf=4 for this plot (Fig. 5d). Because of the catch-bond effect,
. On the basis of our experiments, we used the median value nf=4 for this plot (Fig. 5d). Because of the catch-bond effect,  decreases from 280 to 22 μm−2; for f >3pN, the velocity becomes independent of force. According to this plot, the minimum density of catch-bond motor per accessible filament to form tubes corresponds to about 10 myo1b bound to 100 nm radius endosomes. This quantity is in the same range as the number of proteins that have been measured at the surface of a synaptic vesicle30. In conclusion, our theoretical model accounts for our experimental data and highlights the importance of myo1b catch-bond property for its in vivo function.
decreases from 280 to 22 μm−2; for f >3pN, the velocity becomes independent of force. According to this plot, the minimum density of catch-bond motor per accessible filament to form tubes corresponds to about 10 myo1b bound to 100 nm radius endosomes. This quantity is in the same range as the number of proteins that have been measured at the surface of a synaptic vesicle30. In conclusion, our theoretical model accounts for our experimental data and highlights the importance of myo1b catch-bond property for its in vivo function.
Discussion
Our results demonstrate for the first time that a myosin-1 has the capacity to tubulate membrane along F-actin bundles at physiological density due to its catch-bond behaviour. Membrane-pulling processes rely on the specific mechanical properties of the motors. For processive kinesins, less than 10 motors at the tip are involved in tube pulling against forces up to 30 pN31, and growth occurs at low motor density on the membrane. In contrast, membrane tubulation by the non-processive kinesin NCD requires a high density of motors to pull tubes against the same range of forces9,25,26, whereas myo1b pulls at low motor density (see phase diagram, Fig. 5b). In addition, NCD motors exhibit periods of backward motion while tubes pulled by myo1b advance at constant velocity always in the same direction. Our theoretical model predicts that these differences are due to myo1b catch-bond properties. According to this model, the formation of stochastic clusters on the tube and at its tip is not required for myo1b unlike for NCD25,26, and the number of myosins at the tip should be small (less than 10). A possible future challenge will be to investigate whether other classes of motor with different mechanical properties can pull membrane tubes. KIF1A is an interesting candidate32: it is very inefficient as a single molecule but can overcome much higher force than conventional kinesin when self-organized in clusters, and thus being well-adjusted to pulling membrane tubes against high load33.
Our observations indicate that bundling of F-actin facilitates tube pulling by myo1b, by decreasing the threshold density of motors on the membrane. This raises the question regarding the organization of the F-actin network (branched or bundled) available in the TGN region in vivo. A population of short and branched F-actin, revealed by a tropomyosin isoform, was observed close to Golgi membranes by electron microscopy34; moreover, Arp2/3-dependent F-actin structures are associated with the base of membrane tubules that exit from the TGN13. Nevertheless, the formin mDia1, which stimulates actin nucleation and actin bundle elongation, participates also in the formation of carrier vesicles in this region35. Both actin organizations being present at the TGN, the next question will be therefore to identify the organization used by myo1b in this region.
In cells, both microtubule-associated motors and myosins are involved in membrane tubule formation and elongation from the trans-Golgi network4,5,6,13. We propose that myo1b acts prior to microtubule-associated motors in the formation of tubular-carrier precursors. Myo1b and F-actin being closely associated to the membrane compartment may first pull small tubes that will then reach microtubules and allow kinesins to relay myosins for tubule elongation. Myosins and F-actin would thus bridge the gap between cell membranes and microtubules.
Methods
Reagents
Alexa Fluor 488 5-SDP ester, Alexa Fluor 594 phalloidin, Texas Red 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (Texas Red DHPE), Alexa Fluor 647 conjugated actin from rabbit muscle, non-labelled phalloidin and streptavidin-coated bead were purchased from Life Technologies. LabSafe GEL Blue was from G-Biosciences. Non-labelled actin from rabbit skeletal muscle was purchased from Cytoskeleton. L-α-phosphatidylcholine from the chicken egg (egg PC), L-α-phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2) from the porcine brain, cholesterol and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[biotinyl(polyethylene glycol)-2000] (DSPE-PEG-biotin) were purchased from Avanti Polar Lipids. Sigillum wax was from Vitrex Medical A/S. B-PER reagent was from Pierce Chemical. Glutathione Sepharose beads, thrombin, and Superdex 200 13/300GL column were purchased from GE Healthcare. Protein A gold was kindly provided by Dr Graça Raposo (Institut Curie, UMR 144). A myo1b plasmid and an anti-myo1b antibody were prepared as described in ref. 13. Other chemicals were purchased from Sigma Aldrich.
Myo1b purification and fluorescent labelling
Myo1b was purified from HEK293-Flp-In cells expressing FLAG-myo1b according to ref. 13. More precisely, HEK293-Flp-In cells expressing FLAG-myo1b were cultured in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum and 0.18 mg ml−1 hygromycine in a spinner flask at 37 °C under 5% CO2, and collected by centrifugation (1,000 g, 10 min, 4 °C) to obtain a 4–5 g of cell pellet. The pellet was lysed in 15 ml of buffer A (30 mM HEPES at pH 7.5, 150 mM KCl, 4 mM MgCl2, 1 mM EGTA, 4 mM ATP, 1 mM dithiothreitol (DTT), 0.1% protease inhibitor cocktail (PIC), 1% Triton X-100) for 30 min at 4 °C and centrifuged at 3,400 g for 10 min at 4 °C. The collected supernatant was then ultracentrifuged (250,000 g, 60 min, 4 °C). The solution between pellet and floating lipid layer was carefully harvested and incubated with 150 μl of anti-FLAG beads for 2 h at 4 °C. The beads were collected by centrifugation (1,000 g, 5 min, 4 °C), washed three times with buffer A and washed once with buffer B (buffer A without Triton X-100 supplemented with 0.1% methylcellulose). FLAG-myo1b was then eluted from the beads by incubating with 0.24 mg ml−1 of 3X FLAG peptide in 300 μl buffer B for 3 h at 4 °C. After removal of the beads by centrifugation (1,000 g, 3 min, 4 °C), the protein solution was dialysed against buffer C (buffer B with 1 mM ATP) overnight at 4 °C to remove the 3 × FLAG peptide.
For the in vitro tube extraction experiments, myo1b was fluorescently labelled using Alexa Fluor 488 5-SDP ester. Two microlitres of 10 mM Alexa Fluor 488 5-SDP ester in dimethyl sulphoxide was added to the protein solution after the dialysis and incubated for 8 h at 4 °C, then the solution was dialysed against buffer C overnight at 4 °C to remove free fluorescent probe. After snap freezing, the protein solution was kept at −80 °C and used during 1–2 months.
The motor activity of purified and fluorescently labelled myo1b was confirmed by using F-actin gliding assay, which gave an average velocity of 56.0±4.3 nm s−1 (Supplementary Fig. 2).
GUV and small liposome preparation
GUVs were grown from lipid films composed of 52% L-α-phosphatidylcholine from the chicken egg (Egg PC), 28% cholesterol, 20% L-α-phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2) and 0.3% Texas Red 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (Texas Red DHPE) (mol/mol) by using the electroformation technique on ITO slides in 256 mOsm sucrose solution. With this method, we cannot predict the amount of PI(4,5)P2 eventually incorporated into the GUVs24. To ensure to work at saturation of the charged density, we worked with more than 8% (mol/mol) PI(4,5)P2 in our lipid film24. We obtained the largest number of GUVs binding myo1b with GUVs made from lipid films containing 20% PI(4,5)P2 (mol/mol). The obtained GUV solution was diluted 10-fold with buffer D (30 mM HEPES at pH 7.5, 100 mM KCl, 2 mM MgCl2, 1 mM EGTA, 1 mM DTT, 2 mM ATP and 0.1 mg ml−1 β-casein) supplemented with glucose to adjust the osmolarity to 258 mOsm. Using optical tweezers, the bending rigidity of the GUV membrane with this composition was measured to be 31.7±3.0 kBT (Supplementary Note and Supplementary Fig. 8). Small liposomes of the same composition as GUVs except Texas Red DHPE were prepared at 1 mg ml−1 lipid concentration by first drying a lipid film formed from a lipid solution and by vortexing buffer D without β-casein for 5 min in a glass vial. Small liposomes were used immediately after preparation.
Cryo-electron microscopy observation
Purified myo1b bound to small liposomes and purified myo1b fraction were analysed by cryo-EM. Binding of myo1b to small liposomes was achieved by incubating 200 μg ml−1 of purified myo1b with small liposomes at 0.5 mg ml−1 lipid concentration for 15 min at 4 °C before flash-freezing. A glow-discharged Quantifoil R1.2/1.3 copper grid (Quantifoil) was loaded with 5 μl of sample solution that was subsequently blotted and vitrified in liquid ethane by plunge freezing. Images were recorded under low-dose conditions with a FEI CM120 operating at 120 kV with a SSD GATAN 1k × 1k camera (Gatan Inc.) at a magnification of 35,000 resulting in 4.96 Å per pixel. The intended underfocus was set to −1.5 to −2 μm. For immunogold labelling, myo1b solution containing rare native vesicles was diluted down to 40 μg ml−1 with 30 mM HEPES at pH 7.5, 100 mM KCl and 2 mM MgCl2, and incubated with 1% bovine serum albumin (BSA) and anti-FLAG antibody at 20-fold dilution for 1 h at RT. Next, Protein A gold was added at 20-fold dilution. After 30 min incubation, sample was flash-frozen for cryo-EM observation.
In vitro tube extraction experiments
Human GST-fascin was expressed in E. coli36. After lysis of bacteria in phosphate buffer containing B-PER reagent GST-fascin was isolated using Glutathione Sepharose beads. Fascin was collected from the beads using 80 units of thrombin and further purified over the Superdex 200 13/300GL column. Two micromolars of Alexa Fluor 647 actin and 1.5 μM fascin were incubated in buffer E (10 mM imidazole-HCl at pH 7.5, 100 mM KCl, 1 mM MgCl2, 1 mM EGTA, 2 mM ATP) for 2 h at RT to form fluorescent actin/fascin bundles. Phalloidin in methanol was added at the final concentration of 24 μM and incubated for 1 h at RT to stabilize the bundles. Fluorescently labelled myo1b at 200 nM in buffer D, corresponding to 258 mOsm, and GUV solution were mixed and incubated for 30 min at 4 °C. An observation chamber was built by placing a 5-mm-thick polydimethylsiloxane block with a 6 mm diameter hole on a poly-L-lysine-coated glass slide. Forty-three microlitres of fluorescent actin/fascin bundle solution was introduced in the chamber and incubated for 10 min at RT. After removal of the bundle solution, the chamber was incubated with 100 μl of buffer D supplemented with 0.4 mg ml−1 β-casein for 5 min at RT to passivate the surface. The chamber was rinsed twice with 100 μl of buffer D. Finally, the mixture of GUV and myo1b was added to the chamber and observed at RT. Fluorescent myo1b, GUV and actin/fascin bundle were visualized through a × 63 oil immersion objective with a confocal microscope (LSM 510 Meta, Carl Zeiss). Note that quantitative parameters can be deduced from only a few % of the GUVs for the following reasons. (1) In all, 10–20% of GUVs are covered with myo1b, probably due to heterogeneity in PI(4,5)P2 distribution. (2) In this fraction, we observe tubes arising from 10% of myo1b-covered GUVs only. (3) Only tubes that we observe growing are analysed, to be sure that they were pulled by myo1b and not due to other factors (flow in the chamber for instance).
Myo1b density on tubes and tube diameter measurements
The myo1b density on a membrane tube ρ and the tube diameter dtube were estimated from the fluorescence signal of the confocal microscopy images. The fluorescence intensities of myo1b (Imyo1b) and lipid (Ilipid) on a tube were measured after background signal subtraction. The number of myo1b molecules (nmyo1b) corresponding to Imyo1b was estimated as follows. The concentration of original myo1b solution was determined by using a BCA assay. Dilutions of the original solution were prepared at eight different concentrations (Cmyo1b). Samples containing solutions of myo1b at these dilutions were scanned by confocal microscopy at a fixed scan volume V=(scan area) × (scan depth) at three different horizontal positions and at the same height in the chamber. Parameters of the confocal microscope were set to be the same as the tube pulling experiments. The average of the fluorescence intensities of each dilution, Imyo1b, was plotted versus the number of myo1b molecules, nmyo1b, contained in the volume (nmyo1b=Cmyo1b × NA × V where NA is Avogadro constant). From the plot, we calibrate the number of myo1b from the fluorescence intensity nmyo1b=α × Imyo1b and measured the α constant. We then calculated nmyo1b on a tube from Imyo1b in the tube region. Next, the correlation between Ilipid and the membrane surface area (Slipid) was calculated as follows. Twenty GUVs were scanned by confocal microscopy in the same conditions as the tube pulling experiments, providing circular cross-sectional images of GUVs. Each Slipid was calculated from the contour length of the GUV image multiplied by the scan depth. The β constant was deduced from the average of 20 GUVs according to Slipid=β × Ilipid.
The myo1b density on the tube was then derived from Imyo1b and Ilipid in the tube region using  . The tube diameter was derived for a tube of length l as
. The tube diameter was derived for a tube of length l as  . Note that all tube diameters used when measuring motor density were smaller than the scan depth, which was typically 0.8 μm.
. Note that all tube diameters used when measuring motor density were smaller than the scan depth, which was typically 0.8 μm.
Sequential images of fluorescent lipids were taken every 10 or 30 s during the extraction for monitoring the dynamics; the last lipid image, together with myo1b and actin fluorescence, was used for the quantitative measurements. We checked whether the bleaching of fluorescent lipids was negligible in tubes during the recording growth time. Three representative bleaching curves obtained from tube regions in different GUVs are shown in Supplementary Fig. 9. We excluded the effect of bleaching of the lipid fluorescence since the bleaching is not significant.
Membrane bending rigidity measurement
A GUV with the same composition as that used in the rest of the experiments, supplemented with 0.1% (mol/mol) DSPE-PEG-biotin was partially aspirated in a micropipette. A membrane tube was extracted from the opposite side of the GUV by using a streptavidin-coated bead trapped by optical tweezers37. The force exerted by the bead on the membrane is given by  , where κ is the membrane bending rigidity and σ is the membrane tension27. Thus, knowing f and σ, one can deduce κ. σ was set through the micropipette aspiration pressure ΔP, that is, the hydrostatic pressure difference between the experimental chamber and inside of the micropipette38. Application of Laplace law gives
, where κ is the membrane bending rigidity and σ is the membrane tension27. Thus, knowing f and σ, one can deduce κ. σ was set through the micropipette aspiration pressure ΔP, that is, the hydrostatic pressure difference between the experimental chamber and inside of the micropipette38. Application of Laplace law gives  , where Rp and Rv are the pipette and GUV radius, respectively. σ was increased stepwise, and, for each step, f was calculated from the average bead displacement x−x0 from its equilibrium position x0 in the optical trap using the linear relationship f=k(x−x0), where k is the trap stiffness. The bead position was measured by video tracking, and k was determined by using the viscous drag method39. A linear fit of the data then allowed us to determine the bending rigidity κ of the GUV membrane. Supplementary Fig. 8 shows a representative data set during such a measurement. We obtained the value κ=31.7±3.0 kBT (n=9). The error corresponds to s.d.
, where Rp and Rv are the pipette and GUV radius, respectively. σ was increased stepwise, and, for each step, f was calculated from the average bead displacement x−x0 from its equilibrium position x0 in the optical trap using the linear relationship f=k(x−x0), where k is the trap stiffness. The bead position was measured by video tracking, and k was determined by using the viscous drag method39. A linear fit of the data then allowed us to determine the bending rigidity κ of the GUV membrane. Supplementary Fig. 8 shows a representative data set during such a measurement. We obtained the value κ=31.7±3.0 kBT (n=9). The error corresponds to s.d.
Additional information
How to cite this article: Yamada, A. et al. Catch-bond behaviour facilitates membrane tubulation by non-processive myosin 1b. Nat. Commun. 5:3624 doi: 10.1038/ncomms4624 (2014).
References
Soldati, T. & Schliwa, M. Powering membrane traffic in endocytosis and recycling. Nat. Rev. Mol. Cell. Biol. 7, 897–908 (2006).
Vale, R. D. The molecular motor toolbox for intracellular transport. Cell 112, 467–480 (2003).
Anitei, M. & Hoflack, B. Bridging membrane and cytoskeleton dynamics in the secretory and endocytic pathways. Nat. Cell Biol. 14, 11–19 (2012).
Dabora, S. L. & Sheetz, M. P. The microtubule-dependent formation of a tubulovesicular network with characteristics of the ER from cultured cell extracts. Cell 54, 27–35 (1988).
Allan, V. J. & Vale, R. D. Cell cycle control of microtubule-based membrane transport and tubule formation in vitro. J. Cell Biol. 113, 347–359 (1991).
Fullerton, A. T., Bau, M. Y., Conrad, P. A. & Bloom, G. S. In vitro reconstitution of microtubule plus end-directed, GTPgammaS-sensitive motility of Golgi membranes. Mol. Biol. Cell 9, 2699–2714 (1998).
Roux, A. et al. A minimal system allowing tubulation with molecular motors pulling on giant liposomes. Proc. Natl Acad. Sci. USA. 99, 5394–5399 (2002).
Koster, G., VanDuijn, M., Hofs, B. & Dogterom, M. Membrane tube formation from giant vesicles by dynamic association of motor proteins. Proc. Natl Acad. Sci. USA 100, 15583–15588 (2003).
Leduc, C. et al. Cooperative extraction of membrane nanotubes by molecular motors. Proc. Natl Acad. Sci. USA 101, 17096–17101 (2004).
Roland, J. T., Kenworthy, A. K., Peranen, J., Caplan, S. & Goldenring, J. R. Myosin Vb interacts with Rab8a on a tubular network containing EHD1 and EHD3. Mol. Biol. Cell 18, 2828–2837 (2007).
Chibalina, M. V., Seaman, M. N., Miller, C. C., Kendrick-Jones, J. & Buss, F. Myosin VI and its interacting protein LMTK2 regulate tubule formation and transport to the endocytic recycling compartment. J. Cell Sci. 120, 4278–4288 (2007).
Salas-Cortes, L. et al. Myosin Ib modulates the morphology and the protein transport within multi-vesicular sorting endosomes. J. Cell Sci. 118, 4823–4832 (2005).
Almeida, C. G. et al. Myosin 1b promotes the formation of post-Golgi carriers by regulating actin assembly and membrane remodelling at the trans-Golgi network. Nat. Cell Biol. 13, 779–789 (2011).
Hokanson, D. E., Laakso, J. M., Lin, T., Sept, D. & Ostap, E. M. Myo1c binds phosphoinositides through a putative pleckstrin homology domain. Mol. Biol. Cell 17, 4856–4865 (2006).
Komaba, S. & Coluccio, L. M. Localization of Myosin 1b to actin protrusions requires phosphoinositide binding. J. Biol. Chem. 285, 27686–27693 (2010).
Coluccio, L. M. & Geeves, M. A. Transient kinetic analysis of the 130-kDa myosin I (MYR-1 gene product) from rat liver. A myosin I designed for maintenance of tension? J. Biol. Chem. 274, 21575–21580 (1999).
Laakso, J. M., Lewis, J. H., Shuman, H. & Ostap, E. M. Myosin I can act as a molecular force sensor. Science 321, 133–136 (2008).
Batters, C. et al. Myo1c is designed for the adaptation response in the inner ear. EMBO J. 23, 1433–1440 (2004).
Coppin, C. M., Pierce, D. W., Hsu, L. & Vale, R. D. The load dependence of kinesin's mechanical cycle. Proc. Natl Acad. Sci. USA 94, 8539–8544 (1997).
Stafford, W. F., Walker, M. L., Trinick, J. A. & Coluccio, L. M. Mammalian class I myosin, Myo1b, is monomeric and cross-links actin filaments as determined by hydrodynamic studies and electron microscopy. Biophys. J. 88, 384–391 (2005).
Lewis, J. H., Greenberg, M. J., Laakso, J. M., Shuman, H. & Ostap, E. M. Calcium regulation of myosin-I tension sensing. Biophys. J. 102, 2799–2807 (2012).
Lin, T., Tang, N. & Ostap, E. M. Biochemical and motile properties of Myo1b splice isoforms. J. Biol. Chem. 280, 41562–41567 (2005).
Levental, I. et al. Calcium-dependent lateral organization in phosphatidylinositol 4,5-bisphosphate (PIP2)- and cholesterol-containing monolayers. Biochemistry 48, 8241–8248 (2009).
Carvalho, K., Ramos, L., Roy, C. & Picart, C. Giant unilamellar vesicles containing phosphatidylinositol(4,5)bisphosphate: characterization and functionality. Biophys. J. 95, 4348–4360 (2008).
Shaklee, P. M. et al. Bidirectional membrane tube dynamics driven by nonprocessive motors. Proc. Natl Acad. Sci. USA 105, 7993–7997 (2008).
Shaklee, P. M., Bourel-Bonnet, L., Dogterom, M. & Schmidt, T. Nonprocessive motor dynamics at the microtubule membrane tube interface. Biophys. J. 98, 93–100 (2010).
Derenyi, I., Julicher, F. & Prost, J. Formation and interaction of membrane tubes. Phys. Rev. Lett. 88, 238101 (2002).
Thomas, W. E., Vogel, V. & Sokurenko, E. Biophysics of catch bonds. Annu. Rev. Biophys. 37, 399–416 (2008).
Haviv, L., Gov, N., Ideses, Y. & Bernheim-Groswasser, A. Thickness distribution of actin bundles in vitro. Eur. Biophys. J. 37, 447–454 (2008).
Takamori, S. et al. Molecular anatomy of a trafficking organelle. Cell 127, 831–846 (2006).
Campas, O., Kafri, Y., Zeldovich, K. B., Casademunt, J. & Joanny, J. F. Collective dynamics of interacting molecular motors. Phys. Rev. Lett. 97, 038101 (2006).
Okada, Y., Higuchi, H. & Hirokawa, N. Processivity of the single-headed kinesin KIF1A through biased binding to tubulin. Nature 424, 574–577 (2003).
Brugues, J. & Casademunt, J. Self-organization and cooperativity of weakly coupled molecular motors under unequal loading. Phys. Rev. Lett. 102, 118104 (2009).
Percival, J. M. et al. Targeting of a tropomyosin isoform to short microfilaments associated with the Golgi complex. Mol. Biol. Cell 15, 268–280 (2004).
Zilberman, Y. et al. Involvement of the Rho-mDia1 pathway in the regulation of Golgi complex architecture and dynamics. Mol. Biol. Cell 22, 2900–2911 (2011).
Vignjevic, D. et al. Formation of filopodia-like bundles in vitro from a dendritic network. J. Cell Biol. 160, 951–962 (2003).
Sorre, B. et al. Curvature-driven lipid sorting needs proximity to a demixing point and is aided by proteins. Proc. Natl Acad. Sci. USA 106, 5622–5626 (2009).
Kwok, R. & Evans, E. Thermoelasticity of large lecithin bilayer vesicles. Biophys. J. 35, 637–652 (1981).
Cuvelier, D., Derenyi, I., Bassereau, P. & Nassoy, P. Coalescence of membrane tethers: experiments, theory, and applications. Biophys. J. 88, 2714–2726 (2005).
Acknowledgements
We thank C.G. Almeida and M.-T. Prospéri for fruitful discussions and technical advice. We thank F. Waharte and the BioImaging Cell and Tissue Core Facility of the Institut Curie (PICT-IBiSA) for technical support. We thank J. Manzi for providing fascin and P. Martin for his help for gliding assays. We thank C. Leduc, O. Campàs and B. Goud for critical reading of the manuscript. This work has been supported by Institut Curie, Centre National de la Recherche Scientifique (CNRS) and the Agence Nationale pour la Recherche (grant ANR- 09-0027-03). E.C. and P.B.’s groups belong to the CNRS consortium CellTiss, to the Labex CelTisPhyBio (ANR-11-LABX0038) and to Paris Sciences et Lettres (ANR-10-IDEX-0001_02).
Author information
Authors and Affiliations
Contributions
E.C. and P.B. conceived the project. A.Y. generated and analysed the data of reconstituted in vitro experiments. A.M. and J.-F.J. provided the theoretical model and analysis. J.L.-T.-W. contributed to the F-actin gliding assay. D.L. and A.D.C. contributed to the biochemical characterization of myo1b and performed cryo-electron microscopy observations. C.P. performed the measurement of lipid membrane bending rigidity. A.Y., A.M., D.L., J.-F.J., E.C. and P.B. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-10, Supplementary Note 1 and Supplementary References (PDF 2165 kb)
Supplementary Movie 1
Tube extraction from a GUV by myo1b. The movie shows the sequence of confocal microscopy images of lipid fluorescence during the tube extension. This movie corresponds to Figs. 3A-B. (AVI 166 kb)
Supplementary Movie 2
Slow tube extraction from a GUV by myo1b at low density. The movie shows the sequence of confocal microscopy images of lipid fluorescence taken every 10 sec. Motor density on the tube was measured equal to 510 molecules.μm-2. A single tube was formed. Image size is 41 μm × 41 μm. (AVI 1754 kb)
Supplementary Movie 3
Membrane fluctuations observed during a tube extraction event. The movie shows the sequence of confocal microscopy images of lipid fluorescence taken every 30 sec. Myo1b density on the tube was measured equal to 750 molecules.μm-2. Large membrane fluctuations were observed at the top middle of the GUV. Image size is 59 μm × 59 μm. (AVI 385 kb)
Rights and permissions
About this article
Cite this article
Yamada, A., Mamane, A., Lee-Tin-Wah, J. et al. Catch-bond behaviour facilitates membrane tubulation by non-processive myosin 1b. Nat Commun 5, 3624 (2014). https://doi.org/10.1038/ncomms4624
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms4624
This article is cited by
-
Myosin 1b is an actin depolymerase
Nature Communications (2019)
-
Compressive three-dimensional super-resolution microscopy with speckle-saturated fluorescence excitation
Nature Communications (2019)
-
Myosin 1b and F-actin are involved in the control of secretory granule biogenesis
Scientific Reports (2017)
-
Force Generation by Membrane-Associated Myosin-I
Scientific Reports (2016)
-
Building endocytic pits without clathrin
Nature Reviews Molecular Cell Biology (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
