
Abstract
In all metallofullerenes known before this work, metal atoms form single highly polar bonds with non-metal atoms in endohedral cluster. This is rather surprising for titanium taking into account the diversity of organotitanium compounds. Here we show that the arc-discharge synthesis of mixed titanium-lutetium metallofullerenes in the presence of ammonia, melamine or methane unexpectedly results in the formation of TiLu2C@Ih-C80 with an icosahedral Ih(7) carbon cage. Single-crystal X-ray diffraction and spectroscopic studies of the compound reveal an unprecedented endohedral cluster with a μ3-carbido ligand and Ti-C double bond. The Ti(IV) in TiLu2C@Ih-C80 can be reversibly reduced to the Ti(III) state. The Ti=C bonding and Ti-localized lowest unoccupied molecular orbital in TiLu2C@Ih-C80 bear a certain resemblance to titanium alkylidenes. TiLu2C@Ih-C80 is the first metallofullerene with a multiple bond between a metal and the central, non-metal atom of the endohedral cluster.
Similar content being viewed by others
Introduction
One of the unique properties of fullerenes is their ability to encapsulate metal atoms with the formation of endohedral metallofullerenes (EMFs)1,2,3,4. In EMFs, the metals donate their valence electrons to the carbon cage and adopt large positive charges. Therefore, when two or more metal atoms are encapsulated in one fullerene molecule, they experience strong Coulomb repulsion. This destabilizing interaction can be balanced by the formation of clusterfullerenes5, which are EMFs whose endohedral clusters also include non-metals (for example, N, (ref. 6) O, (ref. 7) S, (ref. 8) C2 (ref. 9)). So far, most clusterfullerenes are based on the trivalent metals such as Sc, Y and lanthanides. Thus, the formal charge distribution in the molecules of the most abundant class of EMFs, nitride clusterfullerenes (NCFs) M3N@C2n, can be described as (M3+)3N3−@C2n6− (ref. 10). The frontier molecular orbitals (MOs) in NCFs are usually localized on the carbon cage, and hence the metal atoms in NCFs are inert and their trivalent state is not amenable to variation by chemical or electrochemical means11,12.
The notable exceptions are Ti-based NCFs, TiSc2N@Ih-C80 and TiY2N@Ih-C80 (refs 13, 14, 15). The nitride cluster works as a template to enforce a trivalent state of an endohedral Ti atom, which can be then oxidized or reduced to the TiIV and TiII states13. In addition to the Ti-NCFs, the only known Ti-EMFs are Ti@C28 (seen only in the gas phase so far)16, the carbides Ti2C2@C78 and Ti2C2@C82 (refs 17, 18), and a sulphide Ti2S@C78 (ref. 19).
In all Ti-EMFs known before this work, Ti formed single highly polar bonds with non-metal atoms in the clusters. For example, the intracluster bonding in Ti2C2@C78 can be described as Ti–C≡C–Ti. Thus, the bonding mode of Ti in EMFs is not different from Sc, Y and lanthanides, which is rather surprising taking into account a diversity of organotitanium compounds20,21. In addition to titanocenes, Ti is able to form single, double and triple Ti˙˙˙C bonds. Since the invention of the Ziegler–Natta process of olefin polymerization, many Ti-based organometallic complexes have been used as catalysts of industrial importance. For instance, compounds with Ti=C double bonds of the general formula L2Ti=CR2 (where L is a ligand and R is H or alkyl) known as Schrock carbenes or alkylidenes are widely used in olefination reactions22,23,24,25,26. Ti in L2Ti=CR2 is usually a 16-electron system with LUMO localized on the metal. Due to such an electronic structure, L2Ti=CR2 molecules are very unstable in the pristine form and readily coordinate other groups to Ti or decompose27. As a result, (Cp)2Ti=CR2 species are frequently formed only in situ as transient intermediates28,29. For example, the archetypical Schrock carbene (Cp)2Ti=CH2 is usually obtained from the Tebbe reagent (Cp)2TiCH2(Cl)Al(CH3)2 (ref. 30), Grubbs’ titanocyclobutane31, or Petasis reagent (Cp)2Ti(CH3)2 (ref. 32). So far, (Cp)2Ti=CH2 was detected only by mass-spectrometry33, whereas X2Ti=CH2 (X=H, F, Cl) species were isolated in low-temperature inert gas matrices34,35. At the same time, Ti alkylidenes with a higher coordination number, when Ti-based LUMO is further coordinated, can form stable molecules22,23,36,37,38,39.
In this work we report on a new type of Ti-EMFs which have a double Ti=C bond and μ3-carbido ligand in the TiLu2C cluster. Although Ti here is a 16-electron system with Ti-localized LUMO similar to the (Cp)2Ti=CR2 species, the structure is stabilized by encapsulation of the TiLu2C cluster in the C80 carbon cage.
Results and Discussion
Synthesis of TiLu2C81
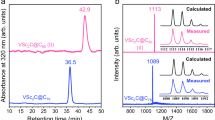
The title compound was first obtained in the arc-discharge synthesis with the use of Lu2O3/Ti2O3 and NH3 as a reactive gas. Under these conditions, the formation of empty fullerenes and conventional EMFs is suppressed yielding Lu3N@C2n NCFs as the main fullerene products (see Fig. 1a). The mixed-metal Ti-Lu EMF was obtained as a minor product, however, its mass-spectrum was different from the expected nitride clusterfullerene TiLu2N@C80 and the composition was identified as TiLu2C81. Similar results were obtained when melamine (C3N6H6) was used as the solid source of nitrogen instead of gaseous NH3 (Fig. 1b). Melamine also partially suppresses formation of empty fullerenes and leads to NCFs as the main products. In contrast to the NH3-assisted synthesis, melamine also resulted in the formation of appreciable amounts of Lu2@C82 and Lu2C2@C82. TiLu2N@C80 was not formed at all, and TiLu2C81 was the main Ti-containing EMF produced. In both the NH3- and the melamine-directed syntheses, TiLu2C81 is the main component of the fraction isolated at a retention time of 35.6–37.5 min. On the basis of chromatographic peak areas, the relative yield of TiLu2C81 versus Lu3N@Ih-C80 in the synthesis with NH3 and melamine is estimated as 1 and 3%, respectively.

(a) synthesis with NH3, (b) synthesis with melamine, (c) synthesis with molecular nitrogen, (d) synthesis with methane as a reactive gas. The arrow indicates the fraction containing TiLu2C81, the red asterisk marks the fraction containing TiLu2N@C80.
In the view of the unexpected formation of TiLu2C81 and the absence of TiLu2N@C80 in the conditions favourable for the synthesis of NCFs, arc synthesis was performed in the presence of molecular nitrogen under conditions used by Yang et al. to synthesize TiSc2N@C80 and TiY2N@C80 (refs 14, 15). This procedure indeed resulted in the formation of TiLu2N@C80, but TiLu2C81 was also found among the products (Fig. 1c). Since TiLu2C81 does not contain nitrogen, the synthesis was also performed in the absence of any nitrogen source with a He atmosphere. However, no TiLu2C81 was detected in such conditions. The best method for the synthesis of TiLu2C81 was found to be a reactive atmosphere method with methane such as the method used in our group earlier to synthesize Sc3CH@C80 (ref. 40). Arc-discharge synthesis in the presence of CH4 yields TiLu2C81 as one of the main EMF products (Fig. 1d) along with Lu3N@Ih-C80 (formed presumably because of the traces of nitrogen in methane and generator) and some Lu2@C2n and Lu2C2@C2n compounds. Isolation of pure TiLu2C81 in the methane-based synthesis can be accomplished in one high-performance liquid chromatography (HPLC) step, since TiLu2C81 is the main component of the fraction eluted at 36–38 min.
Molecular structure of TiLu2C@Ih-C80
The isolation of pure TiLu2C81 (see Fig. 2a for its mass-spectrum) enabled the elucidation of its molecular structure. Two lines in a 3:1 intensity ratio in the 13C NMR spectrum of TiLu2C81 at 136.82 and 143.67 p.p.m. (Fig. 2b) unambiguously show that this endohedral metallofullerene utilizes the icosahedral Ih(7)-C80 carbon cage with a freely rotating endohedral cluster (for comparison, the 13C lines of Lu2ScN@Ih-C80 are found at 137.12 and 143.99 p.p.m.41). Thus, the formula of TiLu2C81 can be written as TiLu2C@Ih-C80. Unfortunately, even after accumulation of the spectrum over two weeks, the signal of the central carbon atom could not be observed.

(a) LDI mass-spectrum of TiLu2C@Ih-C80 with experimental and calculated isotopic distribution; (b) 13C NMR spectrum of TiLu2C@Ih-C80; (c) schematic description of the electronic distribution in TiLu2C@Ih-C80. exp., experimental; LDI, laser desorption ionization; theor., theoretical.
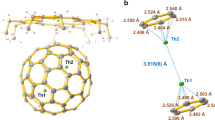
The molecular structure of TiLu2C@Ih-C80 was obtained by single-crystal X-ray diffraction as shown in Fig. 3. The asymmetric unit of TiLu2C@Ih-C80·Ni(OEP)·2(toluene) has no crystallographically imposed symmetry and consists of independent molecules of each of the four components. A typical host–guest interaction occurs between the fullerene and the porphyrin. Also, the two toluenes exhibit face-to-face interaction with the fullerene. The shortest contact between the central Ni atom of the host porphyrin and the cage is Ni1…C1, 2.871(5) Å. The existence of a central, single carbide (C81) was indicated by the increase in agreement factors when C is replaced by N (R1 increases from 0.0578 to 0.0580). Further indications stem from the overall geometric features. Four orientations for TiLu2 were refined. Their relative orientations are shown in Supplementary Fig. 1a. The major orientation of the TiLu2 unit has a fractional occupancy of 0.6475(15). Occupancies of three other minor sites are 0.1747(12), 0.1126(12) and 0.0651(12). Positional disorder in the central carbon position (C81) was not detected but it is likely to exist in correlation with the minor three groups.

(a) A view of the structure of TiLu2C@Ih-C80·Ni(OEP)·2(toluene) with hydrogen atoms omitted for clarity; only the predominant Ti and Lu positions are shown. Displacement parameters are shown at the 50% probability level. (b) The structure of the major TiLu2C cluster with X-ray determined bond lengths and bond angles. The numbers in red are the displacements of the atoms from the plane of the TiLu2C unit. (c) The view showing the interactions of the metal ions with the closest portions of the cage. The dotted circle marked X represents the centre of the C80 cage.
Although there is disorder in the position of the TiLu2 unit, it is fortunate that there is a dominant site with large population. Due to uncertainty in the minor positions, only the major site (65%) for the TiLu2C interior group should be taken as representative of the cluster geometry, and our discussion of the structure will focus on this dominant site. In it, the Ti1–C81 distance is short, at 1.874(6) Å. The Lu–C distances of 2.135(6) and 2.139(6) Å are longer than the Lu–N distances previously found for Lu3N@Ih-C80 (range, 2.00–2.07 Å)42. In TiLu2C@Ih-C80, the trimetallic unit is nearly planar; C81 is 0.076(4) Å from the least-squares plane of the TiLu2C unit. The sum of the angles about C81 is 359.2°. The dihedral angle between the porphyrin plane and the TiLu2C plane is 82.3°. Also, the edge of the TiLu2C plane is not between the two N-Ni-N lines but rather lies approximately parallel to one of them (Supplementary Fig. 1b). This arrangement is in agreement with what is observed in the M3N@Ih-C80 metal porphyrin structures43.
As seen in (c) in Fig. 3, each metal atom has a different interaction with the carbon cage, and those of Ti are particularly short. Notably, C81 is displaced by 0.33 Å from the cage centroid in the direction of Ti1, and Ti1 has the shortest cage contacts. Ti1 is centred on a 5:6 ring junction with Ti1˙˙˙C3 and Ti1˙˙˙C4 distances of 2.137(6) and 2.150(6) Å, respectively. Lu1 is η5 to a pentagon with Lu˙˙˙C distances in the range from 2.309(6) to 2.678(6) Å; Lu2 is η6 to a hexagon with Lu˙˙˙C distances in the range 2.318(5) to 2.563(6) Å. Further evidence for the strong cage interaction with Ti can be seen in the very large pyramidalization angles44 of 13.7° at C3 and C4. Only C67 has a larger pyramidalization angle, 13.8°, corresponding to the shortest Lu1˙˙˙C distance. For comparison, the remaining 77 carbon pyramidalization angles average 9.9° (see Supplementary Fig. 2).
The charge distribution in TiLu2C might be written as a carbide [(Lu3+)2Ti4+C4−]6+, as shown in Fig. 2c. Note that Ti4+C4− fragment is isoelectronic to Sc3+N3−, and hence TiLu2C@Ih-C80 has certain analogy to nitride clusterfullerene Lu2ScN@Ih-C80. However, structural and computational studies and the vibrational spectroscopy discussed below show that a more appropriate description of the cluster structure is [(Lu3+)2Ti3+=C3−]6+ with a double bond between the titanium and the central carbon atom (Fig. 2c). The length of the Ti=C bond in TiLu2C@Ih-C80, 1.874(6) Å, falls into the range of the Ti=C bond lengths in Ti alkylidenes characterized by single-crystal X-ray diffraction (usually 1.8–1.9 Å, see Supplementary Table 1 for a list of values)22,23. The structure of the planar TiLu2C cluster with μ3-carbido ligand bonded to three metal atoms is rather unusual and has only few analogues in organometallic chemistry, and none of them involves Ti45,46,47,48. For instance, Floriani et al. reported the structure in which μ3-C is bonded to two Nb and one Na ions45, whereas Matsuzaka et al. synthesized a Ru2Pt complex with μ3-carbido ligand46. The carbon-metal bonds in these clusters have double bond character and resemble the bonding in corresponding alkylidenes.
DFT calculations of the molecular structure and bonding
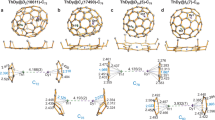
Deeper insight into the molecular and electronic structure of TiLu2C@Ih-C80 was obtained by DFT computations and topological analysis of the electronic density using Quantum Theory of Atoms in Molecules (QTAIM)49. In agreement with the fast rotation of the cluster that was revealed by 13C NMR spectroscopy, computations showed the existence of several orientational conformers of the TiLu2C cluster inside the Ih-C80 cage in the energy range of 0–10 kJ mol−1 (see Supplementary Fig. 3 and Supplementary Table 2). A similar situation is also found for group III nitride clusterfullerenes (for example, Lu2ScN@Ih-C80)10,11,12,41 in contrast to TiM2N@Ih-C80 (M=Sc, Y, Lu), which show more enhanced dependence of the relative energy on the cluster position13,14,15.
The DFT predicted Ti=C bond length in TiLu2C@Ih-C80 is 1.836 Å, ~0.04 Å shorter than experimental value. For comparison, the length of the single Ti–C bond in Ti2C2@C78 predicted at the same level of theory is 1.976 Å (see Table 1 for some other relevant bond lengths).
To learn more about the bonding situation in the TiLu2C cluster as well as the metal–cage bonding, we applied a topological QTAIM analysis. In this work we focus mainly on the QTAIM atomic charges and delocalization indices δ(A,B) which denote the number of electron pairs shared between the atoms A and B. For non-polar bonds, δ(A,B) values have the physical meaning of bond orders in Lewis definition, whereas for polar bonds, the values are reduced compared with the formal bond order. The sum of all δ(M, C) indices for a given metal atom M and carbon atoms from the carbon cage defines a ‘metal-cage bond order’50. Table 1 compares selected QTAIM parameters for clusterfullerenes TiLu2C@Ih-C80, TiLu2N@Ih-C80, Lu2ScN@Ih-C80, Ti2C2@C78, as well as methylidenes L2Ti=CH2 (L=Cp, Cl, and F). An analysis of the bond critical point indicators (electron density, Laplacian, energy densities and so on) is given in Supplementary Table 3.
Depending on the ligand L, the DFT-optimized Ti=C bond lengths in these L2Ti=CH2 species vary from 1.849 Å (Cl) to 1.951 Å (Cp), whereas the δ(Ti, C) values are changing inversely with the bond length from 1.34 (Cp) to 1.71 (Cl). Note that the δ(Ti, C) values are smaller than 2 in all L2Ti=CH2 molecules because of the significant polarity of the Ti=C bond (according to atomic charges, Ti donates 0.60–0.66 e to the carbon atom of the =CH2 group).
QTAIM parameters of the Ti=C bond in TiLu2C@Ih-C80, including the δ(Ti, C) index, atomic charge of Ti, as well as the number of electron pairs shared with the π-ligands, are strikingly similar to those of Cp2Ti=CH2. Besides, the δ(Ti, C) value in TiLu2C@Ih-C80 is approximately two times higher than δ(Sc/Lu, N) values in nitride clusterfullerenes as well as the δ(Ti, C) index for a single Ti–C bond in Ti2C2@C78. These data show that the description of the carbon-titanium bond by a C4−–Ti4+ charge distribution is not appropriate and should be done in terms of double polar C3−=Ti3+ bond. At the same time, the large negative charge of the central carbon atom in TiLu2C@Ih-C80 is very similar to that of the nitrogen atom in NCFs, showing their nucleophilic nature. Thus, QTAIM analysis supports the formal charge distribution depicted in Fig. 2 and clearly shows that the Ti=C bond in TiLu2C@Ih-C80 can be described as a double bond similar to that in Ti alkylidenes (see Supplementary Note 1 and Supplementary Table 3 for further comparison of the Ti–C bond in the TiLu2C cluster and in Ti alkylidenes).
Vibrational spectra
Detailed information on the intracluster as well as metal–cage interactions can be obtained by means of vibrational spectroscopy. In line with the close similarity of their molecular structures, the Fourier transform infrared spectra of Lu2ScN@Ih-C80 (ref. 51) and TiLu2C@Ih-C80 (Fig. 4) exhibit a pronounced similarity in the ranges dominated by the carbon cage vibrations (400–600, 1,000–1,600 cm−1).

Top: room-temperature FTIR spectra on KBr substrate and DFT-computed IR spectrum of TiLu2C@Ih-C80. Bottom: UV–vis-NIR absorption spectra of TiLu2C@Ih-C80 and Lu2ScN@Ih-C80 in toluene solution. calc., calculated; DFT, density dunctional theory; exp., experimental; FTIR, Fourier transform infrared (spectroscopy); NIR, near infrared; UV, ultraviolet.
The main difference between the spectra of two EMFs in the mid-IR range is found for the metal-nitrogen anti-symmetric stretching modes of Lu2ScN@Ih-C80 (denoted as ν(M–N) hereafter) and analogous vibrations of TiLu2C@Ih-C80. In M3N@C80 species, the ν(M–N) mode is two-fold degenerate and corresponds to the motions of the nitrogen atom in the plane of the M3N cluster52. In the mixed-metal nitride cluster such as Lu2ScN, the ν(M–N) mode is split: the higher-frequency component (near 710 cm−1) corresponds to the vibration with the dominant contribution of the Sc–N stretching, whereas the lower-frequency component (at 667 cm−1) has mainly a ν(Lu–N) character51. The lower mass of the carbon atom should result in the 8% upshift of the vibrational frequencies of the ν(M–C) modes in the TiLu2C cluster if the M–C and M–N force constants are equal. However, Lu–C bonds in the TiLu2C@Ih-C80 are noticeably longer than the Lu–N bonds in Lu2ScN@Ih-C80 (2.178 Å versus 2.147 Å in DFT-optimized structure). As the increase of the bond length results in the downshift of its vibrational frequency, the force constant and mass factors in TiLu2C@Ih-C80 compensate, and the ν(Lu–C) mode is found at 670 cm−1, very close to the frequency of the ν(Lu–N) mode in Lu2ScN@Ih-C80. On the contrary, the ν(Ti=C) mode in TiLu2C@Ih-C80 is found at 821 cm−1, which is 16% higher than the frequency of the ν(Sc–N) mode in Lu2ScN@Ih-C80. Obviously, this large shift is caused both by the lower mass of the carbon atom and by a much shorter Ti=C bond in comparison to the Sc−N bond in Lu2ScN@Ih-C80 (1.836 Å versus 1.945 Å). Note also that the ν(Ti=C) frequency in TiLu2C@Ih-C80 is considerably higher than the ν(Ti=C) frequencies of the matrix-isolated F2Ti=CH2 and Cl2Ti=CH2 species found at 706 and 680 cm−1, respectively34,35, which is consistent with the short bond length given by crystal results.
Similar to the IR spectra, the regions of the Raman spectra of two EMFs corresponding to the cage vibrations exhibit a pronounced similarity (Supplementary Fig. 4). For the analysis of the cluster–cage interactions, the most informative one is the low-frequency range of 100–200 cm−1 comprising vibrations of the endohedral clusters (Supplementary Fig. 4).The modes corresponding to the metal–cage interaction are found at approximately the same frequencies both in Lu2ScN@C80 and TiLu2C@C80 showing that cluster–cage interactions are similar in these EMFs in agreement with the same extent of the cluster-to-cage electron transfer (see Supplementary Note 2 for more detailed analysis of the Raman spectra).
Electronic structure of TiLu2C@Ih-C80
Figure 4 compares the absorption spectra of TiLu2C@Ih-C80 and Lu2ScN@Ih-C80 measured in toluene solution. Absorption spectra of EMFs correspond to the π→π* excitations of the carbon cage and hence the spectra of all M2ScN@Ih-C80 species are very similar and closely resemble those of M3N@Ih-C80 (M=Y and lanthanides)51. The absorption onset of Lu2ScN@C80 is observed near 800 nm, and the lowest energy absorption features are distinguished at 666 and 691 nm followed by more intense bands at 558 and 402 nm (Fig. 4, also see ref. 51). In the visible range, the spectrum of TiLu2C@Ih-C80 with the main features at 390 nm and near 540 nm also resembles that of Lu2ScN@Ih-C80, however, the bands are broader and less defined. At the 600–700 nm range, TiLu2C@Ih-C80 exhibits only one broad absorption band at 670 nm (versus two better resolved features in Lu2ScN@Ih-C80). The main difference between the two EMFs is found in the lowest energy range: whereas Lu2ScN@Ih-C80 has no well-defined absorptions beyond 700 nm, TiLu2C@Ih-C80 exhibits one weak band at 910 nm, and its absorption onset is extended to ~1,100 nm. On the basis of the onset and lowest energy absorption, the lowest and highest estimations of the bandgap of TiLu2C@Ih-C80 can be determined as 1.13 and 1.36 eV, respectively, which is ~0.4 eV smaller than analogous values determined for Lu2ScN@Ih-C80 (1.55–1.79 eV). Thus, absorption spectra reveal that in spite of the same electron count and similar nature of the cluster–cage interactions, the electronic properties of TiLu2C@Ih-C80 are noticeably different from those of Lu2ScN@Ih-C80. On the basis of the frontier MO analysis (see below), the lowest energy excitation in TiLu2C@Ih-C80 can be presumably assigned to the cage-to-Ti electron transfer.
Figure 5a compares the results of the cyclic voltammetry and square wave voltammetry studies of TiLu2C@Ih-C80 and Lu2ScN@Ih-C80, and Table 2 lists the measured redox potentials. At room temperature in o-dichlorobenzene (o-DCB) solution, TiLu2C@Ih-C80 exhibits one reversible reduction and one reversible oxidation step at −0.91 and +0.63 V, respectively, (versus the Fc(Cp)2+/0 couple) resulting in an electrochemical gap of 1.54 V.

(a) Cyclic voltammetry and square wave voltammetry (SWV) curves of TiLu2C@Ih-C80 (black lines) and Lu2ScN@Ih-C80 (blue lines); (b) Kohn–Sham MO energy levels (occupied—black, unoccupied—pink) of TiLu2C@Ih-C80, TiLu2N@Ih-C80 and Lu2ScN@Ih-C80; (c) isosurfaces of the frontier MOs of TiLu2C@Ih-C80 and Lu2ScN@Ih-C80.
The reversible reduction of TiLu2C@Ih-C80 is quite remarkable, since NCFs with group III metals (including Lu2ScN@Ih-C80) exhibit electrochemically irreversible reductions2,53. The electrochemical behaviour of TiLu2C@Ih-C80 at the first reduction step is similar to that of other Ti-based EMFs, such as diamagnetic Ti2S@C78, paramagnetic TiSc2N@C80 and TiY2N@C80, which exhibit reversible reductions at −0.92, −0.94 and −1.11 V, respectively, and all EMFs are characterized by the reduction localized at the Ti atom13,15,19. Note also that the Ti(III)/Ti(IV) oxidation potential of Ti-alkylidene (Nacnac)Ti(CH2tBu)2 in THF reported by Mindiola et al. is −0.90 V (Nacnac−=[Ar]NC(Me)CHC(Me)N[Ar], Ar=2,6-(CHMe2)2C6H3) (ref. 37).
The cyclic voltammogram of Lu2ScN@Ih-C80 measured under the same conditions as TiLu2C@Ih-C80 exhibits an irreversible reduction at −1.41 V (peak potential), which is 0.50 V more negative than the first reduction potential of TiLu2C@Ih-C80. On the contrary, the first oxidation of Lu2ScN@Ih-C80 is reversible and occurs at +0.66 V, very close to the oxidation potential of TiLu2C@Ih-C80 (+0.63 V). Interestingly, oxidation potentials of TiM2N@Ih-C80 are found at much more negative values (+0.16 and 0.00 V for M=Sc and Y, respectively). Note also that redox potentials of Lu2ScN@Ih-C80 are close to those of Lu3N@Ih-C80 (ref. 54).
Frontier MO analysis
Experimental studies reveal that the bandgap of TiLu2C@Ih-C80 is 0.4–0.5 eV smaller than that of the isoelectronic Lu2ScN@Ih-C80 because the former has a lower-energy LUMO presumably localized on Ti. On the contrary, HOMO of TiLu2C@Ih-C80 has a similar energy to that of Lu2ScN@Ih-C80. These conclusions are further rationalized by the DFT-based frontier orbital analysis. Figure 5 shows MO energy levels in TiLu2C@Ih-C80, TiLu2N@Ih-C80 and Lu2ScN@Ih-C80 as well as the isosurfaces of the HOMOs and LUMOs. The LUMO of TiLu2C@Ih-C80 is largely localized on the Ti atom resembling that of Cp2Ti=CH2 and is 0.27 eV below the LUMO energy of Lu2ScN@Ih-C80. The latter also has a similar shape to the LUMO of TiLu2C@Ih-C80, but the carbon cage contribution to the MO is much larger at the expense of the lower Sc contribution. The shapes and the energies of the HOMOs of TiLu2C@Ih-C80 and Lu2ScN@Ih-C80 are almost identical, and both MOs are essentially carbon cage orbitals.
MO analysis also explains a similar reductive and different oxidative behaviour of TiLu2C@Ih-C80 and Ti-NCFs. Figure 5 shows that the lowest energy vacant MO of the TiLu2N@C80 at −3.96 eV has almost the same energy as the LUMO of TiLu2C@Ih-C80 (−4.05 eV) and hence their reduction potentials should be similar. On the contrary, oxidation of TiLu2N@Ih-C80 (as well as other TiM2N@Ih-C80) removes one electron from the Ti-based SOMO, whose energy is well above the HOMO energy of TiLu2C@Ih-C80. Hence, oxidation potentials of Ti-NCFs are much lower than that of TiLu2C@Ih-C80. Further details of MO analysis focusing on the intracluster bonding are given in Supplementary Fig. 5 and Supplementary Note 3.
Discussion
In this work we have discovered a new type of endohedral clusterfullerenes with central μ3-C atom and double Ti=C bond. The Ti-C bonding in TiLu2C@Ih-C80 and metal-based LUMO resembles those in the unstable L2Ti=CR2 carbenes with 16-electron Ti (such as Cp2Ti=CH2). However, unlike the latter, TiLu2C@Ih-C80 is stable at the room temperature (in toluene solution kept in the dark, the sample did not show any traces of decomposition over the period of 1.5 years), which can be ascribed to the protective role of the fullerene cage. Finally, TiLu2C@Ih-C80 was fully characterized by spectroscopic and electrochemical techniques and its electronic structure was found to be intermediate between that of conventional MIII-based nitride clusterfullerenes M3N@Ih-C80 and nitride clusterfullerenes with one Ti atom, TiM2N@Ih-C80.
Methods
Synthesis of TiLu2C@C80 with the use of NH3
Mixed-metal cluster fullerenes were produced by evaporating graphite rods in the electric arc in the Krätschmer–Huffman method modified in our group. The graphite rods (length 100 mm, diameter 8 mm) were packed with Lu2O3, Ti2O3, and graphite (molar ratio of Lu:Ti:C=1:1:15) and evaporated in 200 mbar helium atmosphere with addition of ammonia (20 mbar NH3) with the current of 100 A. The soot produced by arc vaporization was first pre-extracted with acetone for one hour to remove non-fullerene products and then Soxhlet extracted for 20 h with carbon disulphide. At the first step, isolation of the fractions containing Lu3N@C2n and TiLu2C@C80 was accomplished by HPLC using analytical 4.6 mm × 250 mm Buckyprep column (Nacalai Tesque, Japan) and toluene as a solvent. The fraction collected at 35.64–37.49 min was then subjected to the second purification step by recycling HPLC at the BuckyPrep column (toluene as eluent, flow rate 1.5 ml min−1). See Supplementary Fig. 6 for HPLC traces and mass-spectrum.
Synthesis of TiLu2C@C80 with the use of other reagents
The synthesis with molecular nitrogen was performed using the same procedure as described for NH3 above, except for the use of an inert He atmosphere (200 mbar) with addition of N2 (20 mbar) instead of NH3 (see Supplementary Fig. 7 for HPLC trace and mass-spectra). The synthesis with melamine was performed using similar procedure, except for the use of an inert He atmosphere (200 mbar) and pure metals (not their oxides). The ratio of the components in the packed rods was Lu:Ti:C:N=1:1:15:10. HPLC traces and mass-spectrum are shown in Supplementary Fig. 8. In the synthesis with methane, the total pressure was 250 mbar (238 mbar He and 12 mbar CH4), the ratio of components in the packed rods was Lu:Ti:C=1:1:12.5.
Spectroscopic measurements
Laser desorption time-of-flight mass-spectra were measured using Biflex III (Bruker, Germany). The 125 MHz 13C NMR spectra were measured at room temperature in carbon disulphide solutions with d6-acetone as a lock on an Avance 500 spectrometer (Bruker, Germany) using the multiprobe head PH 1152Z. Ultraviolet–vis-near-infrared absorption spectra were measured at room temperature in toluene solution on a Shimadzu 3100 spectrophotometer. The Fourier transform infrared spectra were recorded at room temperature in transmission mode by a Vertex 80v spectrometer (Bruker, Germany) with a resolution of 2 cm−1. Raman spectra were recorded at 78 K on a T 64000 triple spectrometer (Jobin Yvon, France) with visible laser radiation (Innova 300 series, Coherent, USA). For IR and Raman measurements, the sample was drop-coated onto single-crystal KBr disks. The residual toluene was removed by heating the polycrystalline films in vacuum at 200 °C for 3 h.
Single-crystal X-ray diffraction
Black blocks were obtained by slow diffusion of a toluene solution of the endohedral fullerene into a toluene solution of Ni(OEP). A crystal suitable for X-ray diffraction was mounted in the nitrogen cold stream at 100(2) K provided by an Oxford Cryostream low-temperature apparatus on the goniometer head of a Bruker D8 diffractometer equipped with an ApexII CCD detector at the Advanced Light Source, Berkeley, California, beamline 11.3.1. Data were collected with the use of silicon(111) monochromated synchrotron radiation (λ=0.77490 Å). The data set was reduced with the use of Bruker SAINT (Bruker AXS Inc., Madison, Wisconsin, USA) and a multi-scan absorption correction applied with the use of SADABS55. The structures were solved by direct methods (SHELXS-2013 (ref. 55)) and refined by full-matrix least-squares on F2 (SHELXL-2013 (ref. 55)). Crystal data: C131H60Lu2N4NiTi, fw=2146.38, triclinic, space group P-1, a=14.6351(7) Å, b=14.6452(7) Å, c=20.4465(10), α=83.295(3), β=83.193(3), γ=60.662(3), V=3784.7(3) Å3, Z=2, 75,518 data, R(int)=0.0568; R1 (24,183 reflections with I>2σ(I))=0.0578, wR2 (all 28,472 data=0.1563, 1,343 parameters, 19 restraints. Further details of crystal refinement are described in Supplementary Note 4.
Electrochemistry
Voltammetric experiments were performed with a PAR 273 potentiostat (EG&G, USA) at room temperature in a glove box. A three-electrode system using a platinum working and a counter electrode and a silver wire reference electrode was used. Potentials were measured by adding ferrocene as an internal standard.
Computations
Optimization of the structures was performed at the PBE56 level using Priroda57,58 package and implemented basis set of TZ2P quality with effective core potential for Ti and Lu atoms. For QTAIM and MO analyses, point energy computations were performed at the PBE level with full-electron TZVP basis set and ZORA correction implemented in ORCA code59,60. QTAIM analysis was performed using AIMAll suite ( http://aim.tkgristmill.com/).
Additional information
Accession codes The X-ray crystallographic coordinates for structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number CCDC 980876. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
How to cite this article: Svitova, A. L. et al. Endohedral fullerene with μ3-carbido ligand and Titanium-Carbon double bond stabilized inside a carbon cage. Nat. Commun. 5:3568 doi: 10.1038/ncomms4568 (2014).
References
Popov, A. A., Yang, S. & Dunsch, L. Endohedral fullerenes. Chem. Rev. 113, 5989–6113 (2013).
Chaur, M. N., Melin, F., Ortiz, A. L. & Echegoyen, L. Chemical, electrochemical, and structural properties of endohedral metallofullerenes. Angew. Chem. Int. Ed. 48, 7514–7538 (2009).
Rodriguez-Fortea, A., Balch, A. L. & Poblet, J. M. Endohedral metallofullerenes: a unique host-guest association. Chem. Soc. Rev. 40, 3551–3563 (2011).
Lu, X., Feng, L., Akasaka, T. & Nagase, S. Current status and future developments of endohedral metallofullerenes. Chem. Soc. Rev. 41, 7723–7760 (2012).
Yang, S., Liu, F., Chen, C., Jiao, M. & Wei, T. Fullerenes encaging metal clusters-clusterfullerenes. Chem. Commun. 47, 11822–11839 (2011).
Zhang, J., Stevenson, S. & Dorn, H. C. Trimetallic nitride template endohedral metallofullerenes: discovery, structural characterization, reactivity, and applications. Acc. Chem. Res. 46, 1548–1557 (2013).
Stevenson, S. et al. A distorted tetrahedral metal oxide cluster inside an icosahedral carbon cage. Synthesis, isolation, and structural characterization of Sc4(μ3-O)2@I h-C80 . J. Am. Chem. Soc. 130, 11844–11845 (2008).
Dunsch, L. et al. Metal sulfide in a C82 fullerene cage: a new form of endohedral clusterfullerenes. J. Am. Chem. Soc. 132, 5413–5421 (2010).
Lu, X., Akasaka, T. & Nagase, S. Carbide cluster metallofullerenes: structure, properties, and possible origin. Acc. Chem. Res. 46, 1627–1635 (2013).
Campanera, J. M., Bo, C., Olmstead, M. M., Balch, A. L. & Poblet, J. M. Bonding within the endohedral fullerenes Sc3N@C78 and Sc3N@C80 as determined by density functional calculations and reexamination of the crystal structure of Sc3N@C78·Co(OEP)·1.5(C6H6)·0.3(CHCl3). J. Phys. Chem. A 106, 12356–12364 (2002).
Popov, A. A. & Dunsch, L. Hindered cluster rotation and 45Sc hyperfine splitting constant in distonoid anion radical Sc3N@C80, and spatial spin charge separation as a general principle for anions of endohedral fullerenes with metal-localized lowest unoccupied molecular orbitals. J. Am. Chem. Soc. 130, 17726–17742 (2008).
Valencia, R. et al. Electronic structure and redox properties of metal nitride endohedral fullerenes M3N@C2n (M=Sc, Y, La, and Gd; 2n=80, 84, 88, 92, 96). Chem.-Eur. J. 15, 10997–11009 (2009).
Popov, A. A., Chen, C., Yang, S., Lipps, F. & Dunsch, L. Spin-flow vibrational spectroscopy of molecules with flexible spin density: electrochemistry, ESR, cluster and spin dynamics, and bonding in TiSc2N@C80 . ACS Nano 4, 4857–4871 (2010).
Yang, S. et al. An endohedral titanium(III) in a clusterfullerene: putting a non-group-III metal nitride into the C80-I h fullerene cage. Chem. Commun. 6391–6393 (2009).
Chen, C. et al. Titanium/Yttrium mixed metal nitride clusterfullerene TiY2N@C80: synthesis, isolation, and effect of the group-III metal. Inorg. Chem. 51, 3039–3045 (2012).
Dunk, P. W. et al. The smallest stable fullerene, M@C28 (M=Ti, Zr, U): stabilization and growth from carbon vapor. J. Am. Chem. Soc. 134, 9380–9389 (2012).
Cao, B. P., Suenaga, K., Okazaki, T. & Shinohara, H. Production, isolation, and EELS characterization of Ti2@C84 dititanium metallofullerenes. J. Phys. Chem. B 106, 9295–9298 (2002).
Cao, B. P. et al. EELS and 13C NMR characterization of pure Ti2@C80 metallofullerene. J. Am. Chem. Soc. 123, 9679–9680 (2001).
Li, F.-F. et al. Ti2S@D3h(24109)-C78: a sulfide cluster metallofullerene containing only transition metals inside the cage. Chem. Sci. 4, 3404–3410 (2013).
Astruc, D. Organometallic Chemistry and Catalysis Springer-Verlag (2007).
Crabtree, R. & Mingos, M. Comprehensive Organometallic Chemistry III. Volume 4: Compounds of Groups 3 to 4 and the f Elements ed Bochmann Manfred Elsevier Ltd (2007).
Mindiola, D. J., Bailey, B. C. & Basuli, F. What a difference one electron makes! generating low-coordinate Ti–C and V–C multiply bonded frameworks through one electron oxidatively induced α-Hydrogen abstractions. Eur. J. Inorg. Chem. 2006, 3135–3146 (2006).
Beckhaus, R. & Santamaría, C. Carbene complexes of titanium group metals— formation and reactivity. J. Organomet. Chem. 617–618, 81–97 (2001).
Beckhaus, R. Carbenoid complexes of electron-deficient transition metals—Syntheses of and with short-lived building blocks. Angew. Chem. Int. Ed. 36, 686–713 (1997).
Schrock, R. R. Transition metal–carbon multiple bonds. J. Chem. Soc., Dalton Trans. 2541–2550 (2001).
Schrock, R. R. High oxidation state multiple metal-carbon bonds. Chem. Rev. 102, 145–180 (2002).
Scott, J. & Mindiola, D. J. A tribute to Frederick Nye Tebbe. Lewis acid stabilized alkylidyne, alkylidene, and imides of 3d early transition metals. Dalton. Trans. 8463–8472 (2009).
Cheon, J., Rogers, D. M. & Girolami, G. S. Mechanistic studies of the thermolysis of tetraneopentyltitanium(IV). 1. Solution evidence that titanium alkylidenes activate saturated hydrocarbons. J. Am. Chem. Soc. 119, 6804–6813 (1997).
Bailey, B. C., Fan, H., Huffman, J. C., Baik, M.-H. & Mindiola, D. J. Intermolecular C–H bond activation reactions promoted by transient titanium alkylidynes. Synthesis, Reactivity, Kinetic, and theoretical studies of the Ti⋮C linkage. J. Am. Chem. Soc. 129, 8781–8793 (2007).
Tebbe, F. N., Parshall, G. W. & Reddy, G. S. Olefin homologation with titanium methylene compounds. J. Am. Chem. Soc. 100, 3611–3613 (1978).
Ott, K. C. & Grubbs, R. H. 1,3-Dimetallacyclobutanes in metal-methylidene dimerization reactions. J. Am. Chem. Soc. 103, 5922–5923 (1981).
Petasis, N. A. & Bzowej, E. I. Titanium-mediated carbonyl olefinations. 1. Methylenations of carbonyl compounds with dimethyltitanocene. J. Am. Chem. Soc. 112, 6392–6394 (1990).
Meurer, E. C., da Rocha, L. L., Pilli, R. A., Eberlin, M. N. & Santos, L. S. Transient intermediates of the Tebbe reagent intercepted and characterized by atmospheric pressure chemical ionization mass spectrometry. Rapid Commun. Mass Spectrom. 20, 2626–2629 (2006).
Lyon, J. T. & Andrews, L. An infrared spectroscopic and theoretical study of group 4 transition metal CH2MCl2 and HC÷MCl3 complexes. Organometallics 26, 332–339 (2006).
Lyon, J. T. & Andrews, L. Group 4 transition metal CH2MF2, CHFMF2, and HC÷MF3 complexes formed by C–F activation and α-fluorine transfer. Inorg. Chem. 46, 4799–4808 (2007).
Bailey, B. C., Huffman, J. C., Mindiola, D. J., Weng, W. & Ozerov, O. V. Remarkably stable titanium complexes containing terminal alkylidene, phosphinidene, and imide functionalities. Organometallics 24, 1390–1393 (2005).
Basuli, F., Bailey, B. C., Tomaszewski, J., Huffman, J. C. & Mindiola, D. J. A terminal and four-coordinate titanium alkylidene prepared by oxidatively induced α-hydrogen abstraction. J. Am. Chem. Soc. 125, 6052–6053 (2003).
Basuli, F. et al. Four-coordinate titanium alkylidene complexes: synthesis, reactivity, and kinetic studies involving the terminal neopentylidene functionality. Organometallics 24, 1886–1906 (2005).
Bailey, B. C., Basuli, F., Huffman, J. C. & Mindiola, D. J. Oxidatively induced α-hydrogen abstraction. A mild protocol to generate terminal titanium alkylidenes containing a β-hydrogen. Organometallics 25, 3963–3968 (2006).
Krause, M., Ziegs, F., Popov, A. A. & Dunsch, L. Entrapped bonded hydrogen in a fullerene: The five-atom cluster Sc3CH in C80 . ChemPhysChem 8, 537–540 (2007).
Yang, S., Popov, A. A. & Dunsch, L. Carbon pyramidalization in fullerene cages induced by the endohedral cluster: non-scandium mixed metal nitride clusterfullerenes. Angew. Chem. Int. Ed. 47, 8196–8200 (2008).
Stevenson, S. et al. Preparation and crystallographic characterization of a new endohedral, Lu3N@C80 · 5 (o-xylene), and comparison with Sc3N@C80 · 5 (o-xylene). Chem.-Eur. J. 8, 4528–4535 (2002).
Stevenson, S., Chancellor, C., Lee, H. M., Olmstead, M. M. & Balch, A. L. Internal and external factors in the structural organization in cocrystals of the mixed-metal endohedrals (GdSc2N@I h-C80, Gd2ScN@I h-C80, and TbSc2N@I h-C80) and nickel(ii) octaethylporphyrin. Inorg. Chem. 47, 1420–1427 (2008).
Haddon, R. C. Pyramidalization - geometrical interpretation of the pi-orbital axis vector in 3 dimensions. J. Phys. Chem. 91, 3719–3720 (1987).
Caselli, A., Solari, E., Scopelliti, R. & Floriani, C. The stepwise four- and six-electron reduction of carbon monoxide to oxyalkylidyne, to carbide and oxide, then to carbide over an Nb−Oxo surface modeled by calix[4]arene1. J. Am. Chem. Soc. 122, 538–539 (2000).
Takemoto, S., Morita, H., Karitani, K., Fujiwara, H. & Matsuzaka, H. A Bimetallic Ru2Pt complex containing a trigonal-planar μ3-carbido ligand: formation, structure, and reactivity relevant to the Fischer−Tropsch process. J. Am. Chem. Soc. 131, 18026–18027 (2009).
Borren, E. S., Hill, A. F., Shang, R., Sharma, M. & Willis, A. C. A golden ring: molecular gold carbido complexes. J. Am. Chem. Soc. 135, 4942–4945 (2013).
Harding, D. J., Kerpal, C., Meijer, G. & Fielicke, A. Unusual bonding in platinum carbido clusters. J. Phys. Chem. Lett. 4, 892–896 (2013).
Bader, R. F. W. Atoms in Molecules—A Quantum Theory Oxford University Press (1990).
Popov, A. A. & Dunsch, L. The bonding situation in endohedral metallofullerenes as studied by Quantum Theory of Atoms In Molecules (QTAIM). Chem.-Eur. J. 15, 9707–9729 (2009).
Yang, S., Popov, A. A., Chen, C. & Dunsch, L. Mixed metal nitride clusterfullerenes in cage isomers: LuxSc3-xN@C80 (x=1, 2) as compared with MxSc3-xN@C80 (M=Er, Dy, Gd, Nd). J. Phys. Chem. C 113, 7616–7623 (2009).
Popov, A. A. Metal-cage bonding, molecular structures and vibrational spectra of endohedral fullerenes: bridging experiment and theory. J. Comput. Theor. Nanosci. 6, 292–317 (2009).
Popov, A. A., Avdoshenko, S. M., Cuniberti, G. & Dunsch, L. Dimerization of radical-anions: nitride clusterfullerenes versus empty fullerenes. J. Phys. Chem. Lett. 2, 1592–1600 (2011).
Chen, N., Pinzón, J. R. & Echegoyen, L. Influence of the encapsulated clusters on the electrochemical behaviour of endohedral fullerene derivatives: comparative study of N-Tritylpyrrolidino derivatives of Sc3N@I h-C80 and Lu3N@I h-C80 . Chemphyschem 12, 1422–1425 (2011).
Sheldrick, G. M. SADABS, SHELXS-2013, SHELXL-2013 University of Göttingen: Germany, (2013).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Laikov, D. N. & Ustynuk, Y. A. PRIRODA-04: a quantum-chemical program suite. New possibilities in the study of molecular systems with the application of parallel computing. Russ. Chem. Bull. 54, 820–826 (2005).
Laikov, D. N. Fast evaluation of density functional exchange-correlation terms using the expansion of the electron density in auxiliary basis sets. Chem. Phys. Lett. 281, 151–156 (1997).
Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2, 73–78 (2012).
Pantazis, D. A. & Neese, F. All-electron scalar relativistic basis sets for the lanthanides. J. Chem. Theory Comput. 5, 2229–2238 (2009).
Elliott, B., Yu, L. & Echegoyen, L. A simple isomeric separation of D5h and I h Sc3N@C80 by selective chemical oxidation. J. Am. Chem. Soc. 127, 10885–10888 (2005).
Chen, N. et al. Comparative spectroscopic and reactivity studies of Sc3-xYxN@C80 (x=0-3). J. Phys. Chem. C 111, 11823–11828 (2007).
Acknowledgements
We are thankful to S. Schiemenz (IFW Dresden) for assistance in spectroscopic measurements and to Dr D. Peryshkin (University of South Carolina) for useful comments on Ti alkylidenes. Financial support from the DFG (grant PO 1602/1-1 to AAP) and the US NSF (Grant CHE-1305125 to A.L.B. and M.M.O.) is gratefully acknowledged. We thank the Advanced Light Source, supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231, for beam time, and Dr Simon J. Teat for his assistance. Computational resources were provided by the Supercomputing Center of Moscow State University and Center for Information Services and High-Performance Computing (ZIH) in TU-Dresden.
Author information
Authors and Affiliations
Contributions
A.A.P., L.D., A.L.B. and M.M.O. designed experiments and wrote the paper. A.L.S., C.S., K.J. and Y.Z. synthesized the samples under supervision of A.A.P. and L.D.; K.B.G., M.M.O. and A.L.B. performed single-crystal X-ray studies; A.A.P. carried out electrochemical measurements and ab initio calculations. All authors discussed the results.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-8, Supplementary Tables 1-3, Supplementary Notes 1-4 and Supplementary References (PDF 1457 kb)
Rights and permissions
About this article
Cite this article
Svitova, A., Ghiassi, K., Schlesier, C. et al. Endohedral fullerene with μ3-carbido ligand and titanium-carbon double bond stabilized inside a carbon cage. Nat Commun 5, 3568 (2014). https://doi.org/10.1038/ncomms4568
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms4568
This article is cited by
-
A stabilization rule for metal carbido cluster bearing μ3-carbido single-atom-ligand encapsulated in carbon cage
Nature Communications (2024)
-
A charged diatomic triple-bonded U≡N species trapped in C82 fullerene cages
Nature Communications (2022)
-
A diuranium carbide cluster stabilized inside a C80 fullerene cage
Nature Communications (2018)
-
Transformation of doped graphite into cluster-encapsulated fullerene cages
Nature Communications (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
