
Abstract
Carbon coating is a simple, effective and common technique for improving the conductivity of active materials in lithium ion batteries. However, carbon coating provides a strong reducing atmosphere and many factors remain unclear concerning the interface nature and underlying interaction mechanism that occurs between carbon and the active materials. Here, we present a size-dependent surface phase change occurring in lithium iron phosphate during the carbon coating process. Intriguingly, nanoscale particles exhibit an extremely high stability during the carbon coating process, whereas microscale particles display a direct visualization of surface phase changes occurring at the interface at elevated temperatures. Our findings provide a comprehensive understanding of the effect of particle size during carbon coating and the interface interaction that occurs on carbon-coated battery material—allowing for further improvement in materials synthesis and manufacturing processes for advanced battery materials.
Similar content being viewed by others

Introduction
Carbon coating is particularly effective for improving conductivity and surface chemistry in specific cathode materials used in lithium ion batteries (LIBs). For example, lithium iron phosphate (LFP) is a promising cathode material with superior stability but its performance is hindered by low electronic conductivity of 10−9 S cm−1 (refs 1, 2, 3); however, an increase of five to eight orders of magnitude in electronic conductivity can be achieved with the use of carbon coating4,5,6,7. Since the first carbon-coating process on LFP was reported by Ravet et al.8,9, numerous studies have been dedicated to understanding the effects of this process10,11,12,13. During carbon coating, the carbon source produces a reducing atmosphere to protect Fe(II) from oxidation at elevated temperatures while simultaneously stabilizing the iron in LFP by hindering its transformation to metallic iron9. Despite significant achievements, a number of issues related to the reducing environment of carbon still remain unclear, including surface/interface reactions that occur between carbon and LFP14; the formation of secondary phases15,16; phase equilibrium17; lithium volatility6; and more importantly, the effect of LFP particle size on these surface/interface reactions18,19,20,21. To answer these questions, a suitable model for LFP material and tools to directly observe the surface/interface reaction are needed. In recent years, the melt-casting process is proposed as a method of choice for the production of industrial class LFP. Ingot LFP has a large flat surface and provides an ideal model to directly study the surface chemistry that occurs during the carbon-coating process22,23,24,25. With the ingot sample design, some basic information such as the interface chemistry reaction that occurs between carbon and LiFePO4 during carbon coating can be obtained.
In this work, LFP ingot material and commercial powder samples are used to demonstrate the direct visualization of a size-dependent surface phase-change process that occurs during the carbon-coating process. Furthermore, this surface phase change can be extended to both micro-sized and larger powder-based LFP materials. Unlike the instability behaviours commonly found in other nanomaterials under high temperatures, in this study, nano-LFP exhibits an extremely high stability during carbon coating. This unusual finding leads to further investigation towards the nature of the interface interaction that occurs between carbon and the underlying material as well as verify the importance of nanosized LFP—consideration of the purity and stability of the material when carbon coating is applied.
Results
Surface new-phase formation
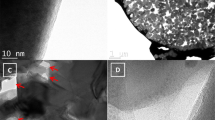
The most intriguing phenomenon is illustrated in Fig. 1. Many ‘island-shaped’ phases (that is, bright colours) are observed on the surface of carbon-coated LFP ingot at 800 °C. These phases clearly arise from the substrate material and were given the name ‘top phase’—so that they may be distinguished from the underlying phase of the substrate. A close-up view outlined in Fig. 1a–d indicates the top phase material consisting of distinguished sharp edges and corners along with the occurrence of a striped pattern. These striped regions range in width from ~100 to 250 nm, and exhibit a three-dimensionally (3D) stripe-structured phase. The island shapes and associated morphological information indicates the formation of a secondary phase. The formation of 3D islands are due to a lattice mismatch-induced stress occurring at the material interface between two phases. During the carbon-coating process, this lattice mismatch stress drives the phase in the form of 3D islands on top of the remaining LFP layer—similar to previous results that mismatch strain evokes the creation of crystal islands26,27,28. We propose that these surface changes are directly related to the introduction of carbon or the carbon-coating process, as this phenomenon is not observed under H2-reducing or Ar atmospheric conditions, where carbon is absent (Supplementary Information).

(a–d) SEM image of surface phase formation on LiFePO4 after carbon coating. (e) Schematic representation of surface phase formation on LiFePO4. Scale bar, 1 mm (a), 500 μm (b), 100 μm (c) and 1 μm (d).
Characterization of surface new phase
Two sample slices—one from the top and the other from the bottom—with a thickness of 50~100 nm were obtained with the use of a focus ion beam (FIB) for observation under a HRTEM (Supplementary Fig. 1). The top sample (Fig. 2a) shows uniform stripes and is consistent with scanning electron microscope (SEM) observations previously mentioned. A uniform carbon layer with a thickness of ~4 nm was also found to coat the surface of the sample (see Fig. 2b and insert in Fig. 2a). Selected Area Electron Diffractions (SAED) in Fig. 2b indicate that the top sample is a new phase corresponding to Fe2P2O7 (Supplementary Fig. 2). In contrast, the bottom phase remains unaltered with no evidence of stripes (Fig. 2c), and is accompanied by a thicker carbon layer (8~10 nm) (Fig. 2d; Supplementary Fig. 3). SAED indicates that this bottom phase remains as LFP. It is evident that carbon coating leads to a surface phase change to occur on flat ingot sample material with the formation of a new secondary phase consistent with Fe2P2O7 (top sample).

(a–d) HRTEM characterization of the top sample (a,b) and bottom sample (c,d). (e) Micro X-ray diffraction (μXRD) patterns and optical images of carbon-coated LiFePO4 ingot at top surface and fresh-cut cross-section. (f) Dynamic secondary ion mass spectrometry (SIMS) profile of C, P, Li and Fe at carbon-coated LiFePO4 ingot. (g) FLY of P L3,2-edge XANES spectra of Fe2P2O7, LiFePO4, and C/LiFePO4 ingot. Scale bar, 1 μm (a,c), 20 nm the insert in panels a and c, 5 nm (b,d) and 5 mm (e).
Micron X-ray diffraction (μ-XRD) was utilized on the carbon-coated sample with the use of two alternate methods—a top view for surface phase analysis and a fresh-cut cross-section for bottom phase identification (Fig. 2e). X-ray patterns of the coated sample surface indicate a large amount of secondary phase Fe2P2O7, whereas the bulk material inside remains as pure LFP. This data further confirms that a phase change occurs only on the sample surface. In contrast, there are no obvious secondary phases found in the XRD pattern for the Ar-annealed sample without carbon precursor additions. The lithium loss and formation of Fe2P2O7 was also confirmed by dynamic secondary ion mass spectrometry and P L3,2-edge X-ray Absorption Near Edge Structure (XANES) spectra29, as shown in Fig. 2f,g.
Synchrotron hard X-ray nano-tomography of surface new phase
The new lithium deficient phase, Fe2P2O7, was also identified with synchrotron hard X-ray full-field spectroscopic imaging and nano-tomography (Fig. 3). The experiment was carried out at X8C beamline30 at the National Synchrotron Light Source, Brookhaven National Laboratory. A cylindrical volume of interest was extracted from the surface of C/LiFePO4 using focused ion beam and lifted-out using micro-manipulation (Fig. 3a,b). The presence of two different phases was confirmed with Fe2P2O7 observed as white (higher X-ray attenuation) at the top of the sample and LiFePO4 being grey at the bottom (Fig. 3c). Two-dimensional chemical mapping (Fig. 3d) and Fe K-edge XANES profile shown across the phase boundary (Fig. 3e) verify the presence of a new phase occurring on the surface of LiFePO4. Three-dimensional tomography was carried out at an X-ray energy of 7,120 eV, where the new phase and LiFePO4 show a distinctive contrast in the Fe K-edge XANES.

(a,b) FIB milling process at the surface of C/LiFePO4 ingot sample. (c) The 3D morphology of the FIB sample. (d) The 2D chemical mapping of selected zone (white dashed box) at the FIB sample. (e) Fe K-edge of XANES profile of selected points in d. Scale bar, 20 μm (a,b), 5 μm (c) and 2 μm (d).
In situ carbon-coating experiment and XRD analysis
We further extended our study to powder-based LFP samples to further validate this interesting phenomenon. Powder-based LFP samples with different sizes (0.5–1 mm, 1–5 μm, 200–500 nm) were obtained by ball milling the above ingot sample. On close inspection of the surface phase-change process, carbon coating was directly observed on powder-based LFP (micron-sized grade, 1~5 μm, Phostech) by in situ SEM observations combined with injection of carbon precursor (ethylene) at 800 °C (Fig. 4a; Supplementary Movie 1). After coating, particle aggregation, shrinkage and surface melting, similar surface phases, form on the surface of LFP (Fig. 4a). During this process, a particularly violent surface-boiling reaction takes place on the larger-sized LFP particles (Supplementary Movie 2). However, following carbon coating, nano-LFP (200–500 nm) shows negligible surface change as well as limited particle aggregation, even under prolonged coating conditions (1 h) (Fig. 4b; Supplementary Fig. 4, Supplementary Movie 3). XRD analysis in Fig. 5a confirms that the new phase only appears with larger LFP samples (1–5 μm, 0.5–1 mm). The size-dependent surface phase change is related to LFP size effect on the carbon decomposition/precipitation process. Coated carbon thickness increases with smaller LFP particle size, as hydrocarbons are readily decomposed on nano-LFP due to enhanced catalytic effects (Supplementary Fig. 5)31.

SEM observation on large-sized (a) and small-sized (b) LiFePO4 powder. Scale bar, 20 μm (a,b).

(a) Micro X-ray diffraction (μXRD) patterns of carbon-coated LiFePO4 with different sizes. (b) A schematic illustration of carbon-competitive diffusion/deposition at different-sized LiFePO4.
Discussion
The formation of new phases was previously reported by Zaghib et al.32 However, new phases of nanosized γ-Fe2O3 clusters were observed for non-carbon-coated LiFePO4, whereas ferromagnetic clusters do not exist in the carbon-coated LiFePO4(ref. 32). During the carbon-coating process, carbon does not penetrate LiFePO4 but rather coats the particle surface. The deposited carbon may result in the reduction of Fe3+, preventing the formation of a ferromagnetic phase. By comparison, an alternate phase and mechanism is observed in our study that may result from the use of different sample size (ranging from nanometres to millimetres) for LiFePO4. Clearly, in our work, carbon coating has an essential role in surface phase change. Supplementary Fig. 6 shows the evolution of surface phase change with carbon coating over time, with increased surface morphology change occurring and larger island-like phases forming. Moreover, the surface phase change is independent of carbon precursor choice with similar changes occurring with various carbon precursors sources (Supplementary Fig. 7). Similar to the decrease in melting point seen for metal and metal oxide materials after carbon introduction, the existence of carbon also decreases the melting point of LFP ingot due to solid solution behaviour. This results in a surface molten process for flat LFP, which is confirmed by the sample being fused to ceramic crucible holder following the carbon-coating process. The decrease in melting point for various metals and metal oxide materials in the presence of carbon has been previously demonstrated33,34. Under the similar conditions, the melting point of Fe2P2O7 is also decreased by carbon coating, although it is theoretically close to 1,200 °C (ref. 35). During this molten process, the volatile and light element, lithium is evaporated in the form of Li2O under the following reaction mechanism:


These phase changes occur readily under moderate reducing environments produced by carbon coating—resulting in a nonstoichiometric mixture existing in the molten phase. During the cooling process, a second phase of Fe2P2O7 precipitates from the nonstoichiometric molten mixture as a result of Li2O loss. These surface phase change occurs under a broad temperature range (Supplementary Fig. 8) and begin to occur at temperatures as low as 650 °C (Supplementary Fig. 9). Our observations are also consistent with results reported by Zaghib et al.36 who designed in situ HRTEM experiments to study the synthesis conditions of C/LiFePO4 for use in LIBs. Their report indicates a structural destruction of LiFePO4 accompanied by the production of unwanted secondary phases occurring at temperatures over 750 °C. Furthermore, heating LiFePO4 to temperatures greater than 800 °C, results in its decomposition. Temperatures lower than 700 °C are not sufficient for the formation of crystalline LiFePO4 and also produces an uneven carbon-coating layer. Therefore, 700 °C is suggested as the optimum synthesis temperature for the production of crystalline LiFePO4 with a homogenous uniform carbon-coating layer. However, their report did not outline in detail the influence of particle size on carbon coating for LiFePO4.
The formation of carbon coating on LFP involves a process of decomposition, deposition and diffusion (Fig. 5b). Following decomposition of carbon precursor, a competitive process ensues between the diffusion of carbon atoms into the crystalline lattice of LFP and aggregation of carbon atoms on the LFP surface. For LFP ingot and larger-sized samples (over several micrometres), hydrocarbon decomposition occurs relatively slowly, resulting in active carbon atoms rapidly submerging into the crystalline lattice of LFP by diffusion before aggregating on the ingot surface. Once the carbon concentration within the LFP reaches a critical value, the melting point of LFP is decreased and the ingot surface melts to form a C/LFP molten alloy. The decrease in melting point and continuous interaction between hydrocarbon and LFP contributes to surface melting, resulting in a molten state and subsequent surface phase reactions occurring. The volatilization of Li2O and/or CO2 and CO from decomposition of carbon precursor forms pockets of gas—as observed in the boiling phenomenon seen under SEM (Supplementary Movie 2). The loss of lithium in large-sized LFP samples was also confirmed by inductively coupled plasma atomic emission spectroscopy (ICP-AES) testing (Supplementary Tables 1 and 2). For nano-LFP particles, however, hydrocarbon decomposition occurs rapidly due to the elevated catalytic effects, resulting in elevated carbon concentration being generated on the particle surface. Furthermore, the precipitation rate of carbon atoms on the surface of nano-LFP is much higher than the diffusion rate of carbon atoms into LFP and a solid carbon layer readily encloses the particle surface before the carbon concentration in the crystal has a chance to reache the critical value required for particle melting. Once nano-LFP is covered with a thick layer of carbon, its catalytic effect ceases and the hydrocarbon decomposition rate decreases substantially—protecting the LFP surface chemistry and limiting the interaction between LFP and hydrocarbon. Thus, nano-LFP exhibits extremely high stability during carbon coating.
In summary, we have shown that a size-dependent surface phase change occurs in LFP during the carbon-coating process. This surface phase change is visible at temperatures as low as 650 °C, which approximates industrial processing temperatures. Our findings show that efforts to achieve a high volumetric energy density for C/LFP through micro and larger-size processes may need to consider the formation of secondary phases, as this phase is non-active and will not contribute to the capacity of LFP. Therefore, the use of nano-C/LFP is a promising strategy for future high-energy density and high-rate capability materials, as they can produce a single phase and stable C/LFP. In addition, it is important to note that such a size-dependent surface phase behaviour may also exist within other olivine phosphate or insulated electrode materials that undergo carbon coating.
Methods
Sample preparation
The LiFePO4 ingot sample was provided by Phostech Lithium Inc. (Montreal, Canada). There are two reasons for choosing this model material: first, the ingot material is commercially available and commonly used, so the typical behaviours are applicable for other LFP materials. Second, this ingot with a flat, smooth and polished surface is ideal for directly observing and investigating surface chemistry change during carbon coating—for both the bulk olivine phase and secondary intercrystalline phases. Melt casting was performed within an air-tight box furnace under an Ar or N2 protection in a graphite or alumina crucible between 950 and 1,050 °C. Low-cost various precursors such as iron, LiOH and H3PO4 were used for ingot synthesis. The detailed precursor compositions have been investigated in a previous paper21. The small laboratory samples (kg grades) were cooled to room temperature within the box furnace under the protection of an Ar atmosphere. The details of the melt-casting process including experimental details, physical property and electrochemical performance can be found in previous work22,23. Here, to get a flat surface on the ingot sample, the melt sample was cut into a smaller size using a manual saw, and then the surface was polished using sandpapers (London, 3M Canada) from coarse (Grit 120) to fine grades (Grit 1500). After ultrasonic cleaning in ethanol several times, a LiFePO4 ingot sample with a clean and flat surface was thus obtained. Powder-based LiFePO4 samples with different sizes (mm, micro and nano grades) were controlled by ball milling the above LiFePO4 ingot sample, and detailed experimental process can be obtained in previous papers24,25. The synthesis technique of LiFePO4 ingot was also described by Zaghib et al.20,36, and a high-rate performance of LiFePO4 was achieved after grinding molten ingot to nanoparticles. In addition, another nano sample, commercial LiFePO4 powder (Phostech) without carbon coating, was also used in this work.
Carbon coating
Carbon-coating experiments on ingot samples were performed in a spray-pyrolysis system, which was previously developed for the synthesis of various nanomaterials. In the experiment, alcohol was used as the carbon precursor and argon acted as a carrying and protecting gas. Briefly, the ingot sample with the flat surface up was put in a quartz tube and Ar was introduced into the quartz tube for 20 min to eliminate the air. Subsequently, the furnace was heated, and the carbon-coating process was performed at temperatures ranging from 600 °C to 900 °C for 20 min. After the carbon coating, the coated sample was cooled to room temperature within the furnace still under the protection of the Ar atmosphere.
Carbon-coated LiFePO4 powder samples were prepared by the conventional solid-state method. Lactose (carbon precursor) was firstly dissolved in either isopropyl alcohol or H2O, and then various LiFePO4 powder samples were added to the carbon precursor solution. The suspension was mixed ultrasonically, and was then allowed to evaporate to dryness. The sample was further annealed at 700 °C for 1 h under Ar protection to form carbon-coated samples.
Characterization
The morphology and microstructure of the samples were characterized using an S-4800 SEM (Hitachi) equipped with an energy-dispersive X-ray microanalysis system and a field-emission gun. X-ray diffraction analyses of the ingot sample was performed using a Bruker D8-Advantage powder diffractometer with Cu K α radiation (1.54056 Å) between 10° and 80° in reflection geometry. Focused ion beam lithography was used for in situ sectioning of the impurity phases, which were imaged and identified by SEM. The slicing process was performed on a LEO (Zeiss) 1540XB FIB/SEM (Western Nanofab., University of Western Ontario), and subsequently the slices were plucked using a nanomanipulator.
After the tip of the end effector was folded over and removed from the nanomanipulator rod, the sample could be mounted in the HRTEM sample holder. HRTEM and energy-dispersive X-ray spectroscopy (EDX) analysis at different phases were performed using a JEOL 2010 FEG instrument operating at an accelerating voltage of 200 kV. The diffraction patterns were also recorded using the SAED mode. The P L3,2-edge XANES spectra were performed at the Canadian Light Source (CLS) on the Variable Line Spacing Plane Grating Monochromator (VLS PGM) beamline. In situ carbon-coating experiment was performed at Hitachi SEM (S-4800) with a heating carrier designed by Protochips, which enable in situ annealing and real-time observation. Carbon gas (ethylene) was introduced to the sample surface in the SEM chamber to perform carbon coating.
Hard X-ray spectroscopic imaging and tomography
The transmission X-ray microscopy spectroscopic imaging and nano-tomography with hard X-ray were conducted at beamline X8C, NSLS, Brookhaven National Laboratories (BNL). A field of view of 40 × 40 μm2 with a 2k × 2k CCD camera was used. A full XANES image series was collected on the FIB sample. Each XANES image series was measured by scanning Fe absorption K-edge from 7,089 to 7,189 eV, with 2 eV step size, and one TXM image at one energy step, which generate 1k × 1k XANES spectra with 2 × 2 binned pixels. Each image was collected with 10 s exposure time. For tomography collection, the data set was collected with 7,120 eV X-rays, using 1,441 projections over an angular range of 180°. The pixel size was 38.9 nm.
All the XANES data analysis was carried out using customized program (MatLab, MathWorks, R2011b) developed in house (beamline X8C group, NSLS, BNL). Background normalization was carried first out for TXM images, with a unique background image collected at every energy. With all 1,024 × 1,024 pixels, we can extract the full spectrum (X-ray intensity versus energy) for each pixel. The standard XANES spectra for the reference materials LiFePO4 (Phostech Lithium Inc.) and new phase were measured at the TXM beamline X8C (NSLS, BNL). The TXM XANES imaging on the reference materials was carried out by using the same condition as used in the XANES imaging for the FIB sample. The tomographic reconstruction was carried out with standard filtered back-projection algorithm.
Additional information
How to cite this article: Wang, J. et al. Size-dependent surface phase change of lithium iron phosphate during carbon coating. Nat. Commun. 5:3415 doi: 10.1038/ncomms4415 (2014).
References
Padhi, A. K., Nanjundaswamy, K. S. & Goodenough, J. B. Phospho‐olivines a positive electrode materials for rechargeable lithium batteries. J. Electrochem. Soc. 144, 1188–1194 (1997).
Goodenough, J. B., Padhi, A. K., Nanjundaswamy, K. S. & Masquelier, C. Cathode materials for secondary (rechargeable) lithium batteries. US Patent 5, 910, 382 (1999).
Malik, R., Burch, D., Bazant, M. & Ceder, G. Particle size dependence of the ionic diffusivity. Nano Lett. 10, 4123–4127 (2010).
Padhi, A. K., Nanjundaswamy, K. S. & Goodenough, J. B. in ECS Meeting Abstract, vol. 96-1 (Los Angeles, CA, 1996).
Striebel, K. et al. LiFePO4/gel/natural graphite cells for the BATT program. J. Power Sources 119–121, 951–954 (2003).
Chung, S. Y., Bloking, J. T. & Chiang, Y. M. Electronically conductive phospho-olivines as lithium storage electrodes. Nat. Mater. 1, 123–128 (2002).
Kang, B. & Ceder, G. Battery materials for ultrafast charging and discharging. Nature 458, 190–193 (2009).
Ravet, N., Magnan, J.-F., Gauthier, M. & Armand, M. in International Conference on Materials for Advanced Technologies (MRS Singapore, 2001).
Ravet, N. et al. Electroactivity of natural and synthetic triphylite. J. Power Sources 97-98, 503–507 (2001).
Herstedt, M. et al. Surface chemistry of carbon-treated LiFePO4 particles for Li-ion battery cathodes studied by PES. Electrochem. Solid State Lett. 6, A202–A206 (2003).
Zhu, C., Yu, Y., Gu, L., Weichert, K. & Maier, J. Electrospinning of highly electroactive carbon‐coated single‐crystalline LiFePO4 nanowires. Angew. Chem. Int. Ed. 50, 6278–6282 (2011).
Chen, Z. & Dahn, J. R. Reducing carbon in LiFePO4/C composite electrodes to maximize specific energy, volumetric energy, and tap density. J. Electrochem. Soc. 149, A1184–A1189 (2002).
Wang, J. J. & Sun, X. L. Understanding and recent development of carbon coating on LiFePO4 cathode materials for lithium-ion batteries. Energy Environ. Sci. 5, 5163–5185 (2012).
Rho, Y. H., Nazar, L. F., Perry, L. & Ryan, D. Surface chemistry of LiFePO4 studied by Mössbauer and X-ray photoelectron spectroscopy and its effect on electrochemical properties. J. Electrochem. Soc. 154, A283–A289 (2007).
Subramanya-Herle, P., Ellis, B., Coombs, N. & Nazar, L. F. Nano-network electronic conduction in iron and nickel olivine phosphates. Nat. Mater. 3, 147–152 (2004).
Salah, A. A., Mauger, A., Julien, C. M. & Gendron, F. Nano-sized impurity phases in relation to the mode of preparation of LiFePO4 . Mater. Sci. Engineer. B 129, 232–244 (2006).
Ong, S. P., Wang, L., Kang, B. & Ceder, G. Li−Fe−P−O2 phase diagram from first principles calculations. Chem. Mater. 20, 1798–1807 (2008).
Wagemaker, M. et al. Dynamic solubility limits in nanosized olivine LiFePO4 . J. Am. Chem. Soc. 133, 10222–10228 (2011).
Gibot, P. et al. Room-temperature single-phase Li insertion/extraction in nanoscale LixFePO4 . Nat. Mater. 7, 741–747 (2008).
Julien, C. M., Mauger, A. & Zaghib, K. Surface effects on electrochemical properties of nano-sized LiFePO4 . J. Mater. Chem. 21, 9955–9968 (2011).
Zaghib, K., Nauger, A., Gendron, F. & Julien, C. M. Surface effects on the physical and electrochemical properties of thin LiFePO4 particles. Chem. Mater. 20, 462–469 (2008).
McNeil, D. D. et al. Melt casting LiFePO4 II. Particle size reduction and electrochemical evaluation. J. Electrochem. Soc. 157, A463–A468 (2010).
Gauthier, M. et al. Melt casting LiFePO4 I. Synthesis and characterization. J. Electrochem. Soc. 157, A453–A462 (2010).
Wang, J. et al. Interaction of carbon coating on LiFePO4: a local visualization study of the influence of impurity phases. Adv. Funct. Mater. 23, 806–814 (2013).
Wang, J. et al. Surface aging at olivine LiFePO4: a direct visual observation of iron dissolution and the protection role of nano-carbon coating. J. Mater. Chem. A 1, 1579–1586 (2013).
Dubrovskii, V. G., Sibirev, N. V., Zhang, X. & Suris, R. A. Stress-driven nucleation of three-dimensional crystal islands: from quantum dots to nanoneedles. Cryst. Growth Des. 10, 3949–3955 (2010).
Makeev, M. A. & Madhukar, A. Calculation of vertical correlation probability in Ge/Si(001) shallow island quantum dot multilayer systems. Nano Lett. 6, 1279–1283 (2006).
Uccelli, E., Arbiol, J., Morante, J. R. & Morral, A. F. InAs quantum dot arrays decorating the facets of GaAs nanowires. ACS Nano 4, 5985–5993 (2010).
Yang, S. et al. Soft X-ray XANES studies of various phases relating to LiFePO4 based cathode materials. Energy Environ. Sci. 5, 7007–7016 (2012).
Wang, J. et al. Automated markerless full field hard x-ray microscopic tomography at sub-50nm 3-dimension spatial resolution. Appl. Phys. Lett. 100, 143107 (2012).
Hoggins, J. T., Swinnea, J. S. & Steinfink, H. Crystal-structure of Fe2P2O7 . J. Solid State Chem. 47, 278–283 (1983).
Salah, A. A. et al. Reduction Fe3+ of impurities in LiFePO4 from pyrolysis of organic precursor used for carbon deposition. J. Electrochem. Soc. 153, A1692–A1701 (2006).
Kuznetsov, V. L., Usoltseva, A. N. & Chuvilin, A. L. Thermodynamic analysis of nucleation of carbon deposits on metal particles and its implications for the growth of carbon nanotubes. Phys. Rev. B 64, 235401 (2001).
Vogel, J. S., Southon, J. R. & Nelson, D. E. Catalyst and binder effects in the use of filamentous graphite for AMS. Nucl. Instrum. Methods Phys. Res. B29, 50–56 (1987).
Baker, R. T. K., Harris, P. S., Thomas, R. B. & Waite, R. J. Formation of filamentous carbon from iron, cobalt and chromium catalyzed decomposition of acetylene. J. Catal. 30, 86–95 (1973).
Trudeau, M. L. et al. In situ high-resolution transmission electron microscopy synthesis observation of nanostructured carbon coated LiFePO4 . J. Power Sources 196, 7383–7394 (2011).
Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC), Clariant Canada (Previous Phostech Lithium Inc.), Canada Research Chair (CRC) Program, Canadian Light Source (CLS), McMaster Microscopy Centre for HRTEM and Western Nanofabrication for FIB, Western University and the MITACS Elevate Strategic Fellowship Program. Use of the NSLS, Brookhaven National Laboratories (BNL) is supported by the US DOE, BES, under contract no. DE-AC02-98CH10886.
Author information
Authors and Affiliations
Contributions
X.S. conceived the overall project; Jiajun W. designed/performed the experiments, data analysis and wrote the manuscript; J.Y. and M.N.B. performed XRD characterization; J.L. helped with in situ carbon-coating experiment; Y.Z. and R.L. helped with TEM characterization and discussion; Y.T. and T.K.S. performed synchrotron soft X-ray characterization and discussion; G.L., M.G. and X.L. interpreted the results; Y.-c.K.C.-W. and Jun W. performed the synchrotron X-ray tomography experiments and data analysis.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-9 and Supplementary Tables 1-2 (PDF 1457 kb)
Supplementary Movie 1
An in-situ SEM movie showing the surface phase formation at microsized LFP particles during carbon coating process. When the temperature increases to 700 oC (around 30 s), LFP surface begins to melt. When it reaches to 800 oC, violent surface melting, aggregation, and shrinkage occur. A new surface phase was formed. The video plays 5 times faster than real time. (AVI 3901 kb)
Supplementary Movie 2
An in-situ SEM movie showing a boiling phenomenon occurring at a larger micro sized LFP particle during carbon coating. During carbon coating at 800 oC, a surface boiling and bubble formation/breaking indicated that surface/interface reaction of C/LFP is particularly violent for larger sized LFP sample. This video plays in real time. (AVI 1480 kb)
Supplementary Movie 3
An in-situ SEM movie showing negligible surface change occurring at nano-LFP particles during carbon coating at 800 oC. This surface phase formation and melting phenomenon does not occur with nano-sized LFP and very little aggregation was found at even longer coating times. The video plays 60 times faster than real time. (AVI 4402 kb)
Rights and permissions
About this article
Cite this article
Wang, J., Yang, J., Tang, Y. et al. Size-dependent surface phase change of lithium iron phosphate during carbon coating. Nat Commun 5, 3415 (2014). https://doi.org/10.1038/ncomms4415
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms4415
This article is cited by
-
Boosting fast energy storage by synergistic engineering of carbon and deficiency
Nature Communications (2020)
-
Impact of pH on preparation of LiFePO4@C cathode materials by a sol-gel route assisted by biomineralization
Ionics (2019)
-
Formation of size-dependent and conductive phase on lithium iron phosphate during carbon coating
Nature Communications (2018)
-
In-situ construction of NiCo2O4 nanoarrays on La0.8Sr0.2MnO3-δ electrodes for intermediate temperature solid oxide fuel cells
Journal of Solid State Electrochemistry (2018)
-
LiFePO4 synthesized via melt synthesis using low-cost iron precursors
Journal of Solid State Electrochemistry (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
