
Abstract
The atmospheric nuclear testing in the 1950s and early 1960s and the burn-up of the SNAP-9A satellite led to large injections of radionuclides into the stratosphere. It is generally accepted that current levels of plutonium and caesium radionuclides in the stratosphere are negligible. Here we show that those radionuclides are present in the stratosphere at higher levels than in the troposphere. The lower content in the troposphere reveals that dry and wet deposition efficiently removes radionuclides within a period of a few weeks to months. Since the stratosphere is thermally stratified and separated from the troposphere by the tropopause, radioactive aerosols remain longer. We estimate a mean residence time for plutonium and caesium radionuclides in the stratosphere of 2.5–5 years. Our results also reveal that strong volcanic eruptions like Eyjafjallajökull in 2010 have an important role in redistributing anthropogenic radionuclides from the stratosphere to the troposphere.
Similar content being viewed by others


Introduction
Anthropogenic radionuclides have been injected into the atmosphere by nuclear weapon tests (NWT) in the fifties, sixties and seventies, the burn-up of the SNAP-9A satellite1, and accidents in nuclear power plants such as Chernobyl (1986) and Fukushima (2011). Radioactive debris became rapidly attached to ambient aerosol particles, which determined the mechanisms governing their atmospheric transport. In the troposphere, dry and wet deposition removes most contaminants within a few weeks to months. In the stratosphere, mean residence time for radioactive debris is longer due to the thermal stratification and separation from the troposphere by the tropopause and the smaller size distribution of the aerosols2. It is generally accepted that the current level of plutonium (Pu) in the stratosphere is negligible and that the tropospheric level is mainly controlled by resuspension of Pu-bearing particles deposited on land3,4,5,6,7,8,9.
There are, however, limited available data on radionuclide transport and distribution in the stratosphere, the size distribution of the radioactive particles and its interaction with natural aerosols. Most information comes from early studies in the sixties and seventies on anthropogenic radionuclides in the atmosphere10,11,12,13,14,15,16,17,18. In these studies, tropospheric and stratospheric aerosols were sampled with the aid of aircraft and balloons. Because of the significant decrease in the atmospheric level of most radionuclides, as a consequence of the introduction of the Nuclear Test Ban Treaty (NTBT) in 1963, most laboratories abandoned stratospheric surveillance in the early eighties. The collected radionuclide data were nonetheless very useful for investigating atmospheric processes19. Results show that the transfer of radioactive debris from the stratosphere to the troposphere takes place mainly at the tropopause breaks, with a maximum in spring and a minimum in autumn13,16,20.
Radioactive particles in the size range of 1–10 μm settle rapidly out of the stratosphere with mean residence times of the order of weeks to months2,12,21. However, the bulk of stratospheric radioactivity was associated with particles below a few tenths of a micrometre in radius (<0.1 μm) and persisted in the stratosphere for years2,20. Radioactive particles with radius below 0.02 μm were typically found above 27 km, and particles of larger radius were found between the tropopause and 21 km. Mean residence times of the order of 1–4 years were reported for radioactive particle aerosols in the stratosphere2,13,15,16,20. These estimations were based on the rates of change of the stratospheric burden over short timescales (2–4 years), and numerous important factors/parameters were not taken into account. We could mention, for example, transport processes in the stratosphere (for example, horizontal mixing, interhemispheric transfer), the longer residence times for small particles (older tail of a residence time distribution) and the effect of stratospheric mixing of debris from different NWTs.
Other studies on Pu radionuclides and 137Cs in the atmosphere are based mainly on monitoring their long-term evolution in surface aerosols and atmospheric depositions. These studies have consistently suggested that Pu activities in the stratospheric compartment may have decayed with mean residence times of 1.0–1.7 years22,23,24,25. The general mixing of stratospheric with tropospheric air is the main mechanism of NWT deposition. However, Pu activity in the stratosphere is not controlled by the same processes as in the troposphere. Several studies have shown that Pu in ground-level air is strongly influenced by resuspension of Pu in soil particles6,7,26,27,28,29. Hence, the estimation of a mean residence time of Pu in the stratosphere by this approach may have been biased to lower values.
High-altitude aerosols (from 8 to 13 km above sea level) have been collected periodically since 1970 under the framework of the environmental radioactivity survey of Switzerland with the aid of aircraft30,31. Elevated concentrations of anthropogenic radionuclides were detected in tropospheric aerosol samples after the passage over Switzerland of the ash plume of the Eyjafjallajökull volcano eruption in 2010. Activities of Pu and 137Cs were, for example, up to three orders of magnitude higher than the levels reported for ground-level aerosols30. These were surprising results and at first glance contradicted previous studies on radionuclides in the atmosphere.
The purpose of this work is to measure Pu, 241Am and 137Cs in air filters from different layers of the troposphere (ground level to upper troposphere) and the lower level of the stratosphere with the aim to determine the residence time of both radionuclides in the atmosphere. We use a 241Am/241Pu age-dating model to determine the time of the introduction of Pu into the atmosphere. In addition, we study the effect of the volcanic eruption of the Eyjafjallajökull volcano of 2010 to reveal the potential role of volcanic eruptions on the redistribution of radioactive contamination present in the atmosphere.
Results
Anthropogenic radionuclides in the atmosphere
The activities of 239,240Pu in stratospheric and tropospheric aerosols over Switzerland are displayed in Table 1. Graphical representation of 239,240Pu and 137Cs in stratospheric and tropospheric aerosols as a function of the year of collection over Switzerland are shown in Figs 1 and 2. 239,240Pu activities in stratospheric aerosols of Switzerland in the seventies were comparable to the activities reported by the Stardust, Airstream and Ashcan high-altitude sampling programmes of the Environmental Measurements Laboratory (Fig. 1)11,14,17,18. 239,240Pu and 137Cs in the stratosphere have since steadily decreased, and current levels are down to two orders of magnitude lower than the ones measured in 1974 (Fig. 1). 239,240Pu activities in stratospheric aerosols are nonetheless up to five orders of magnitude higher than in ground-level aerosols at present days (Fig. 1). 137Cs behaved similarly, with high-altitude aerosols having up to three orders of magnitude higher activities than ground-level aerosols (Fig. 2).

239,240Pu in stratospheric aerosols from Switzerland (red traingles) and in aerosols samples from the high-altitude aerosol monitoring programmes of the Environmental Measurements Laboratory (light blue rhombus): stardust, airstream and ashcan11,14,17,18. 239,240Pu in the Eyjafjallajökull volcano ash plume (orange triangles) and at ground-level air in Switzerland (green rhombus) are also shown. The lines represent the Pu distribution obtained by the exponential model for different mean residence times (τ=1, 2.5, 5 and 10 a). Uncertainties are expressed at 95% level of confidence (k=2).

137Cs in low tropospheric (green rhombus) and stratospheric (red triangles) aerosols of Switzerland. 137Cs in the Eyjafjallajökull volcano ash plume is also shown (orange triangles). The lines represent the 137Cs distribution obtained by the exponential model for different mean residence times (τ=1, 2.5, 5 and 10 a). Uncertainties are expressed at 95% level of confidence (k=2).
Activity and isotopic ratios of radionuclides
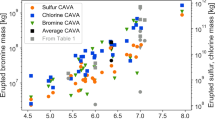
We used the 241Am/241Pu age-dating method32 on selected aerosol samples to corroborate the time of the introduction of Pu in the atmosphere. The calculated dates showed consistently that the Pu contamination occurred between 1964 and 1982 (Table 2). Activity ratios, recalculated to the date of sampling, of 12–14 for 241Pu/239,240Pu and 240Pu/239Pu isotope ratios close to 0.18 (Table 2) are also consistent with the ratios reported in atmospheric aerosols after the NTBT in 1963 (ref. 1). This is corroborated by 241Am/239,240Pu ratios close to 0.5. 238Pu/239,240Pu ratios higher than expected for NWT fallout (typical ratios of 0.02–0.03) suggest additional Pu sources (Fig. 3). Enriched 238Pu in the stratosphere was observed during the years after the burn-up of the 238Pu-powered satellite SNAP-9A at about 50 km in the atmosphere in 1964 (ref. 15). This event increased significantly the amount of 238Pu in the atmosphere (Fig. 3). The relatively high 238Pu/239,240Pu activity ratios observed currently indicates that 238Pu originating mainly from the satellite accident and from NWT with high 238Pu/239,240Pu, possibly injected in the upper stratosphere, is still circulating in the stratosphere. A 239,240Pu/137Cs mean activity ratio of 0.0118 was found in post-moratorium (NTBT) stratospheric aerosols (Fig. 4), which is in good agreement with the average 239,240Pu/137Cs activity ratio of 0.012 reported for global fallout33,34. Nevertheless, recent 239,240Pu/137Cs activity ratios in stratospheric aerosols deviate significantly from the above correlation line (Fig. 4). This may be explained by their different chemical/physical properties (for example, volatilities, particle affinities in the atmosphere and so on).

Activity ratios in stratospheric aerosols from Switzerland (red traingles) and in aerosol samples from the high-altitude aerosol monitoring programmes of the Environmental Measurements Laboratory (light blue triangles): stardust, airstream and ashcan11,14,17,18. The dashed lines at 0.02 and 0.04 represent the current range of 238Pu/239,240Pu activity ratios in ground-level aerosols and soil samples from Switzerland.

Red triangles represent the post-moratorium (after the NTBT in 1963) stratospheric aerosols (1973–1986). Blue triangles represent recent stratospheric samples taken after 2004.The correlation line for the post-moratorium samples is also shown.
Discussion
The determination of radioelement isotopic ratios and the 241Am/241Pu-dating method all confirmed that Pu contamination in the stratosphere was mainly originated from the NWTs fallout and the burn-up of the satellite SNAP-9A. Over the past decades, stratospheric 239,240Pu and 137Cs activities showed a large variability that suggests a low mixing rate of stratospheric air masses and/or the input of these radionuclides from upper layers of the stratosphere. The lower variability observed in ground-level air may indicate that equilibrium or steady-state conditions have been reached for the sources (for example, resuspension, transfer from the stratosphere) and sinks (for example, deposition) of these radionuclides in the troposphere. Our results show that significant fractions of radioactive aerosols (possibly small particles <0.1 μm) remain in the stratosphere for timescales of the order of several decades. Mean residence time of stratospheric air can be obtained from observations of tracer changes in the atmosphere35,36,37,38,39,40,41. We used an exponential distribution model (box model) to interpret the trend of Pu and 137Cs in the stratosphere of Switzerland. This choice was justified by the sparse data set of Pu and 137Cs in stratospheric aerosols, which is not suitable for estimating annual trends. In addition, the distributions of our data sets fit well the asymmetric shape of the exponential distribution, with a peak for short residence times and a long tail for long residence times. The radionuclide is assumed to be distributed homogeneously over the stratospheric domain. The model does not resolve the spatial distribution of the radionuclide within the stratospheric domain, but it describes the activity of the radionuclide (for example, 239,240Pu, 137Cs) inside a box representing the stratosphere over Switzerland. We assumed that horizontal inflow and outflow of the radionuclide into the box are similar. However, vertical downward transport from upper layers of the atmosphere into the stratospheric box and out of the box through the tropopause may have strong effects on the radioisotope activity and therefore on the mean age of the tracer. Best agreement between the measured and modelled Pu activities were obtained for mean residence times of Pu in the stratosphere of 2.5–5 years (Fig. 1). The mean residence times were further confirmed by the 137Cs data (Fig. 2). These results are in good agreement with previous estimations of the mean residence time of stratospheric air using sampling methods like aircrafts, high-altitude balloons and satellite observations35,38,40. Radioisotopes introduced in the atmosphere as a consequence of the nuclear bomb tests during the fifties and sixties are still useful tracers to study transport processes.
The aerosol samples collected in the low tropopause (altitude: 1–3 km) during the Eyjafjallajökull event showed Pu activities and 238Pu/239,240Pu activity ratios similar to the ones observed in stratospheric aerosols (Figs 1 and 3, Table 1). This result indicates that the Pu detected in the volcano ash plume may have come from the stratosphere. The Eyjafjallajökull is a sub-glacial volcano covered by an ice cap of about 77 km2, which is about 250 m thick at the summit of 1,666 m above sea level. During the eruption of April 2010, thousands of tons of molten rock came into contact with ice creating a huge explosion throwing steam and particles high into the air. This explosive eruption introduced volcanic fine-grained ash and gases (for example, SO2) up to the lowermost stratosphere42,43. In the atmosphere, sulphur-bearing gases such as SO2 transformed into tiny sulphuric acid particles2,44. Aerosol size distributions within ash clouds showed a fine mode (0.1–0.6 μm) associated with sulphuric acid and/or sulphate, and a coarse mode (0.6–35 μm) associated with ash45. Numerous studies showed that the Eyjafjallajökull ash particles were strongly depolarizing46 and that particles were electrically charged47,48. Harrison et al.47 proposed radioactive decay as a potential source for this particle depolarization. Charging of aerosols has significant effects on their transport mechanisms like changes in the vertical deposition speeds and particle–particle agglomeration rates49. Charged particles have, for example, increased collision efficiencies and therefore large scavenging coefficients50. We hypothesize that the introduction of ash particles and gases into the stratosphere by the Eyjafjallajökull volcano is responsible for an increased scavenging of Pu and 137Cs aerosols in the stratosphere and an increased transport into the troposphere by sedimentation.
This hypothesis is further supported by the presence of cirrus-like formations over Europe after the volcano eruption. Volcanic ash particles favour ice nucleation (IN). Seifert et al.51 showed the existence of very efficient IN during the Eyjafjallajökull volcanic event. Bingemer et al.52 also observed a large increase of the IN concentration during this event, with IN surface densities of at least a factor two higher than that in other conditions (for example, air masses affected by mineral dust). Seifert et al.51 demonstrated the presence of ash-assisted formation of cirrus-like clouds in the troposphere over Leipzig and Maisach during several days after the first observational period (14–17 April 2010). This was further confirmed by the detection of a volcanic ash-induced cirrus cloud at altitudes of 8–11 km over Jülich during the passage of the Eyjafjallajökull ash plumes46. The backward trajectory analysis confirmed that the cirrus cloud was originated from ash loadings of air masses near Iceland46. The air masses had relative humidities slightly above 100% in which the formation of ice crystals was very favourable. The ash layer then descended with the cirrus cloud. Trickl et al.43 concluded that a branch of the ash plume was present in the tropopause over Garmisch-Partenkirchen on 19 April, and that some volcanic contribution in the lowermost stratosphere cannot be excluded. Backward trajectories revealed Iceland as the closest source region for the air masses of the troposphere and lowermost stratosphere43. Trickl et al.43 suggested that the cirrus formation occurred between 14 and 16 April 2013 in the upper troposphere over Iceland where favourable conditions for aerosol growth existed (relative humidity exceeded 80% several times). Here we show that the strong volcanic eruption of the Eyjafjallajökull volcano has redistributed anthropogenic radionuclides in the lower atmosphere. Waugh38 emphasized the need for more measurements of ‘age tracers’. We believed that NWT radionuclides could be a very good proxy for the study of atmospheric air mass transfer during the last 50 years. In this respect, Pu is the perfect candidate because it has a long physical half-life and a high distribution coefficient on mineral and organic particles, and do not react with atmospheric components under light irradiation.
Methods
Sampling and laboratory methods
High-altitude aerosols were collected six times a year with especially prepared military airplanes. This sampling programme started in the seventies but it was interrupted between the end of the eighties and 2004 (ref. 31). Air volumes between 200 and 3,000 m3 were filtered through cellulose filters (555 × 526 mm2, type 0048, Whatman). The filters were immediately analysed by gamma spectrometry31,53. Ground-level aerosols (at 1 m above the soil surface) were collected with high volume samplers (500–800 m3 h−1) at five different locations of Switzerland. The samples were analysed by gamma spectrometry on a weekly basis.
Since 1994, the cumulative samples of 1 year for each sampling location (annual air volumes >200,000 m3) was analysed for alpha emitters. After the eruption of the Eyjafjallajökull volcano in 2010, aerosol samples were collected during the passage over Switzerland of the ash plume at the altitude of 1–3 km (ref. 30). In this study, we selected several samples of high-altitude aerosol for analysing Pu and 241Am. The air filters were ashed at 550 °C for 48 h before the radiochemical analysis. The radiochemical method combined high-pressure microwave digestion for the dissolution of the ashes with the highly selective extraction chromatographic resins Tetravalent (TEVA, the extractant is a quaternary ammonium salt) and N,N,N′,N′-tetra-n-octyldiglycolamide (DGA) (supplied by Triskem International, Bruz, France) to purify Pu and americium54. The alpha sources were prepared by electrodeposition on stainless steel discs. High-resolution alpha spectrometry was performed on an alpha spectrometer with passivated implanted planar silicon (PIPS) detectors (Alpha Analyst, Canberra Electronic, France). Selected Pu alpha sources were redissolved in concentrated HNO3 and HCl as described in ref. 32 for determining either the activity of 241Pu by liquid scintillation counting (Quantulus 1220, Wallac) or the concentration of 239Pu and 240Pu by sector-field inductively coupled plasma mass spectrometry equipped with a desolvation unit for the reduction of 238U1H molecular interference isobaric to 239Pu. The inductively coupled plasma mass spectrometry analysis was carried out on an Element 2 at the Federal Office of Public Health according to ref. 55.
Age distribution model
We have chosen an exponential distribution model to interpret our radionuclide data in stratospheric air. The response function of the exponential model (g(t)) is represented by

where T represents the mean residence time of the distribution and t is the time. The whole residence time distribution, also named age spectra38, is only characterized by T, which represents in our study case the mean residence time of the radionuclide in the stratosphere. The initial activity of the radionuclide in the stratospheric box is set for the year 1970, a few years after the entry into force of the NTBT. The input of the radionuclide into the stratospheric layer in the seventies is calculated from the activity values reported by the Environmental Measurements Laboratory11,14,17,18. After 1980, any additional input of 239,240Pu is assumed to be negligible.
Additional information
How to cite this article: Corcho Alvarado, J.A. et al. Anthropogenic radionuclides in atmospheric air over Switzerland during the last few decades. Nat. Commun. 5:3030 doi: 10.1038/ncomms4030 (2014).
References
UNSCEAR: Sources and Effects of Ionising Radiation. Report to the General Assembly, Vol. 1, Annex C (United Nations Scientific Committee on the Effects of Atomic Radiation, 2000).
Martell, E. A. The size distribution and interaction of radioactive and natural aerosols in the stratosphere. Tellus XVIII, 486–498 (1966).
Zerle, L. et al. The 41Ca bomb pulse and atmospheric transport of radionuclides. J. Geophys. Res. 102, 19517–19527 (1997).
Warneke, T., Croudace, I. W., Warwick, P. E. & Taylor, R. N. A new ground-level fallout record of uranium and plutonium isotopes for northern temperate latitudes. Earth Planet Sci. Lett. 203, 1047–1057 (2002).
Igarashi, Y., Otsuji-Hatori, M. & Hirose, K. Recent deposition of 90Sr and 137Cs observed in Tsukuba. J. Environ. Radioact. 31, 157–169 (1996).
Hirose, K. et al. Recent trends of plutonium fallout observed in Japan: plutonium as a proxy for desertification. J. Environ. Monitor 5, 302–307 (2003).
Hirose, K., Igarashi, Y. & Aoyama, M. Analysis of the 50-year records of the atmospheric deposition of long-lived radionuclides in Japan. Appl. Radiat. Isotopes 66, 1675–1678 (2008).
Chamizo, E., García-León, M., Enamorado, S. M., Jiménez-Ramos, M. C. & Wacker, L. Measurement of plutonium isotopes, 239Pu and 240Pu, in air-filter samples from Seville (2001- 2002). Atmos. Environ. 44, 1851–1858 (2010).
Povinec, P. P. et al. Long-term variations of 14C and 137Cs in the Bratislava air--implications of different atmospheric transport processes. J. Environ. Radioact. 108, 33–40 (2012).
Feely, H.,W. & Spar, J. Tungsten-185 from nuclear bomb tests as a tracer for stratospheric meteorology. Nature 188, 1062–1064 (1960).
Feely, H. W., Krey, P. W., Spar, J. & Walton, A. The High Altitude Sampling Program Vol. 1, 2A, 2B, 3, 4, and 5. Defense Technical Information Center (1961).
Sisefsky, J. Debris from tests of nuclear weapons. Science 133, 735–740 (1961).
Feely, H. W., Seitz, H., Lagomarsino, R. J. & Biscaye, P. E. Transport and fallout of stratospheric radioactive debris. Tellus 18, 316–328 (1966).
Feely, H. W., Katzman, D., Seitz, H., Davidson, B. & Friend, J. P. Final Report on Project StarDust Report DASA-2166, Defense Atomic Support Agency, Washington, DC, USA (1967).
Telegadas, K. The seasonal stratospheric distribution of plutonium-238 and strontium-90, March through November 1967. HASL-204, 1, I2–16, U.S. Atomic Energy Commission (1969).
List, R. J. & Telegadas, K. Using radioactive tracers to develop a model of the circulation of the stratosphere. J. Atmos. Sci. 26, 1128–1136 (1969).
Leifer, R. & Juzdan, Z. R. The High Altitude Sampling Program: Radioactivity in the Stratosphere - Final Report Report No. EML-458 (Environmental Measurements Laboratory, US Department of Energy, New York, USA (1986).
Leifer, R. Project Airstream: Trace Gas Final Report Report No. EML-549 (Environmental Measurements Laboratory, US Department of Energy, New York, USA (1992).
Machta, L. Meteorological benefits from atmospheric nuclear tests. Health Phys. 82, 635–643 (2002).
Allkofer, O. C. & Fox, J. M. New conceptions on the meteorology of stratospheric fallout from nuclear weapon tests. Arch. Meteorol. Geophys. Bioklimatol. A 15, 299–317 (1966).
Edvarson, K., Löw, K. & Sisefsky, J. Fractionation phenomena in nuclear weapons debris. Nature 184, 1771–1774 (1959).
Miyake, Y., Katsuragi, Y. & Sugimura, Y. Deposition of plutonium in Tokyo through the end of 1966. Papers Meteorol. Geophys. 19, 267–276 (1968).
Lee, S. C. et al. The origin of plutonium in the atmosphere. Geochem. J. 19, 283–288 (1985).
Hirose, K. & Sugimura, Y. Plutonium isotopes in the surface air in Japan: effect of Chernobyl accident. J. Radioanal. Nucl. Chem. 138, 127–138 (1990).
Hirose, K., Igarashi, Y., Aoyama, M. & Miyao, T. inRadioactivity in the Environment Vol. 1, ed. Kudo A. 251–266Elsevier (2001).
Igarashi, Y., Aoyama, M., Hirose, K., Miyao, T. & Yabuki, S. Is it possible to use 90Sr and 137Cs as tracers for the aeolian dust transport? Water Air Soil Poll. 130, 349–354 (2001).
Rosner, G. & Winkler, R. Long-term variation (1986-1998) of post-Chernobyl 90Sr, 137Cs, 238Pu and 239,240Pu concentrations in air, depositions to ground, resuspension factors and resuspension rates in south Germany. Sci. Total Environ. 273, 11–25 (2001).
Igarashi, Y. et al. Resuspension: decadal monitoring time series of the anthropogenic radioactivity deposition in Japan. J. Radiat. Res. 44, 319–328 (2003).
Hölgye, Z. Plutonium isotopes in surface air of Prague in 1986-2006. J. Environ. Radioact. 99, 1653–1655 (2008).
Müller, M. & Estier, S. Überwachung der Radioaktivität der Luft mit Militärflugzeugen. Environmental Radioactivity and Radiation Exposure in Switzerland - Annual Report Federal Office of Public Health: Switzerland, (2010).
Völkle, H. Überwachung der Radioaktivität der Luft mit Militärflugzeugen. Environmental Radioactivity and Radiation Exposure in Switzerland - Annual Report Federal Office of Public Health: Switzerland, (2006).
Corcho Alvarado, J. A., Chawla, F. & Froidevaux, P. Determining Pu-241 in environmental samples: case studies in alpine soils. Radiochim. Acta 99, 121–129 (2011).
Koide, M., Michel, R., Goldberg, E. D., Herron, M. M. & Langway, C. C. Jr Depositional history of artificial radionuclides in the Ross Ice Shelf, Antarctica. Earth Planet Sci. Lett. 44, 205–223 (1979).
Koide, M., Michel, R., Goldberg, E. D., Herron, M. M. & Langway, C. C. Jr Characterization of radioactive fallout from pre- and post-moratorium tests to polar ice caps. Nature 296, 544–547 (1982).
Waugh, D. W. & Hall, T. M. Age of stratospheric air: theory, observations, and models. Rev. Geophys. 40, 1-1–1-26 (2002).
Andrews, A. E. et al. Mean ages of stratospheric air derived from in situ observations of CO2, CH4, and N2O. J. Geophys. Res. 106, 32295–32314 (2001).
Plumb, R. A. Stratospheric Transport. J. Meteor. Soc. Jpn 80, 793–809 (2002).
Waugh, D. Atmospheric dynamics: the age of stratospheric air. Nat. Geosci. 2, 14–16 (2009).
Engel, A. et al. Age of stratospheric air unchanged within uncertainties over the past 30 years. Nat. Geosci. 2, 28–31 (2009).
Stiller, G. P. et al. Observed temporal evolution of global mean age of stratospheric air for the 2002 to 2010 period. Atmos. Chem. Phys. 12, 3311–3331 (2012).
Boering, K. A. et al. Stratospheric mean ages and transport rates from observations of CO2 and N2O. Science 274, 1340–1343 (1996).
Gislason, S. R. et al. Characterization of Eyjafjallajökull volcanic ash particles and a protocol for rapid risk assessment. Proc. Natl Acad. Sci. 108, 7307–7312 (2011).
Trickl, T., Giehl, H., Jäger, H. & Vogelmann, H. 35 yr of stratospheric aerosol measurements at Garmisch-Partenkirchen: from Fuego to Eyjafjallajökull, and beyond. Atmos. Chem. Phys. 13, 5205–5225 (2013).
Ansmann, A. et al. The 16 April 2010 major volcanic ash plume over central Europe: EARLINET lidar and AERONET photometer observations at Leipzig and Munich, Germany. Geophys. Res. Lett. 37, L13810 (2010).
Johnson, B. et al. In situ observations of volcanic ash clouds from the FAAM aircraft during the eruption of Eyjafjallajökull in 2010. J. Geophys. Res. 117, D00U24 (2012).
Rolf, C., Krämer, M., Schiller, C., Hildebrandt, M. & Riese, M. Lidar observation and model simulation of a volcanic-ash-induced cirrus cloud during the Eyjafjallajökull eruption. Atmos. Chem. Phys. 12, 10281–10294 (2012).
Harrison, R. G., Nicoll, K. A., Ulanowski, Z. & Mather, T. A. Self-charging of the Eyjafjallajökull volcanic ash plume. Env. Res. Lett. 5, 024004 (2010).
Ansmann, A. et al. Ash and fine-mode particle mass profiles from EARLINET-AERONET observations over central Europe after the eruptions of the Eyjafjallajökull volcano in 2010. J. Geophys. Res. 116, D00U02 (2011).
Ulanowski, Z., Bailey, J., Lucas, P. W., Hough, J. H. & Hirst, E. Alignment of atmospheric mineral dust due to electric field. Atmos. Chem. Phys. 7, 6161–6173 (2007).
Tripathi, S. N. & Harrison, R. G. Scavenging of electrified radioactive aerosol. Atmos. Env. 35, 5817–5821 (2001).
Seifert, P. et al. Ice formation in ash-influenced clouds after the eruption of the Eyjafjallajökull volcano in April 2010. J. Geophys. Res. 116, D00U04 (2011).
Bingemer, H. et al. Atmospheric ice nuclei in the Eyjafjallajökull volcanic ash plume. Atmos. Chem. Phys. Discuss. 11, 2733–2748 (2011).
Winiger, P., Hubek, 0, Halter, J. & Michaud, B. Konzentrationsmessungen von Be-7, Cs-137 und jungen Spaltfragmenten an der Tropopause. Tellus XXVIII5, 434–441 (1976).
Luisier, F., Corcho Alvarado, J. A., Steinmann, P. h., Krächler, M. & Froidevaux, P. A new method for the determination at ultra-low levels of plutonium and americium, using high pressure microwave digestion and alpha-spectrometry or ICP-SMS. J. Radioanal. Nucl. Chem. 281, 425–432 (2009).
Froidevaux, P. & Haldimann, M. Plutonium from above-ground nuclear tests in milk teeth: investigation of placental transfer in children born between 1951 and 1995 in Switzerland. Environ. Health Perspect. 116, 1731–1734 (2008).
Acknowledgements
We acknowledge the financial support of the Swiss Federal Office of Public Health. We also acknowledge the long-term support of the Swiss Air Force for taking the high-altitude aerosol samples.
Author information
Authors and Affiliations
Contributions
P.S., P.F. and J.A.C.A. organized the research. P.S. and S.E. provided the samples and performed gamma spectrometry analysis. J.A.C.A. and P.F. performed the radiochemical analyses. M.H. was in charge of the ICP-MS analysis. P.S., P.F., F.B. and J.A.C.A. interpreted the data. P.S., P.F. and J.A.C.A. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Alvarado, J., Steinmann, P., Estier, S. et al. Anthropogenic radionuclides in atmospheric air over Switzerland during the last few decades. Nat Commun 5, 3030 (2014). https://doi.org/10.1038/ncomms4030
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms4030
This article is cited by
-
Environmental radioactivity measurements in soil using inductively coupled plasma mass spectrometry and gamma-ray spectrometry in various areas in Cameroon
Journal of Radioanalytical and Nuclear Chemistry (2023)
-
Characteristics of radioactivity in the surface air along the 45°N zonal belt in South-Eastern Europe
International Journal of Environmental Science and Technology (2022)
-
Fracturing and healing of basaltic magmas during explosive volcanic eruptions
Nature Geoscience (2021)
-
Atmospheric fallout of radionuclides in peat bogs in the Western Segment of the Russian Arctic
Environmental Science and Pollution Research (2021)
-
Pre-Chernobyl deposition dynamics of anthropogenic radionuclides at Cluj-Napoca, Romania
Journal of Radioanalytical and Nuclear Chemistry (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
