
Abstract
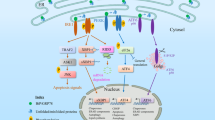
Nitric oxide (NO) is a pleiotropic signalling molecule that binds to cytochrome c oxidase (complex IV) reversibly and in competition with oxygen1,2,3. This action of NO has both physiological and pathophysiological consequences. Here we report that endogenously generated NO, which disrupts the respiratory chain, may cause changes in mitochondrial calcium flux. This induces cleavage of the endoplasmic reticulum (ER) stress-regulated transcription factor p90 ATF6 into an active p50 form. Cleavage depends on a calcium-dependent serine protease through a regulated intramembrane proteolysis (RIP) process4,5. p50 ATF6 then translocates to the nucleus to upregulate expression of the ER-resident molecular chaperone, glucose-regulated protein 78 (Grp78)4. The increase in Grp78 provides significant cytoprotection6 against toxic agents, including thapsigargin, a selective ER calcium–ATPase inhibitor7. Cytoprotection is abolished after treatment with cyclosporin A (CsA), which disrupts mitochondrial calcium signalling8, or with the calcium chelator BAPTA-AM9. The NO-mediated ER stress response is diminished in rho0 cells devoid of mitochondrial DNA10, consistent with our evidence that NO-dependent mitochondrial disruption is coupled to the ER stress response.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


References
Moncada, S. & Erusalimsky, J. D. Does nitric oxide modulate mitochondrial energy generation and apoptosis? Nature Rev. Mol. Biol. 3, 214–220 (2002).
Brown, G. C. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim. Biophys. Acta 1504, 46–57 (2001).
Cooper, C. E. Nitric oxide and cytochrome oxidase: substrate, inhibitor or effector? Trends Biochem. Sci. 27, 33–39 (2002).
Ye, J. et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 6, 1355–1364 (2000).
Brown, M. S., Ye, J., Rawson, R. B, & Goldstein, J. L. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell 100, 391–398 (2000).
Reddy, R. K. et al. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. J. Biol. Chem. 278, 20915–20924 (2003).
Thastrup, O., Cullen, P. J., Drobak, B. K., Hanley, M. R. & Dawson, A. P. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc. Natl Acad. Sci. USA 87, 2466–2470 (1990).
Crompton, M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 341, 233–249 (1999).
Chen, L. Y., Chiang, A. S., Hung, J. J., Hung, H.l., & Lai, Y. K. Thapsigargin-induced grp78 expression is mediated by the increase of cytosolic free calcium in 9L rat brain tumor cells. J. Cell Biochem. 78, 404–416 (2000).
King, M. P. & Attardi, G. Isolation of human cell lines lacking mitochondrial DNA. Methods Enzymol. 264, 304–313 (1996).
Xu, W., Liu, L., Smith, G. C. & Charles, I. G. Nitric oxide upregulates expression of DNA-PKcs to protect cells from DNA-damaging antitumour agents. Nature Cell Biol. 2, 339–345 (2000).
Xu, W., Liu, L. & Charles, I. G. Microencapsulated iNOS-expressing cells cause tumor suppression in mice. FASEB J. 16, 213–215 (2002).
Packer, M. A. & Murphy, M. P. Peroxynitrite causes calcium efflux from mitochondria which is prevented by cyclosporin A. FEBS Lett. 346, 237–240 (1994).
Horn, T. F. et al. Nitric oxide promotes intracellular calcium release from mitochondria in striatal neurons. FASEB J. 16, 1611–1622 (2002).
Schweizer, M. & Richter C. Peroxynitrite stimulates the pyridine nucleotide-linked Ca2+ release from intact rat liver mitochondria. Biochemistry 35, 4524–4528 (1996).
Yoshida, H. et al. Endoplasmic reticulum stress-induced formation of transcription factor complex ERSF including NF-Y (CBF) and activating transcription factors 6α and 6b that activates the mammalian unfolded protein response. Mol. Cell. Biol. 21, 1239–1248 (2001).
Gotoh, T., Oyadomari, S., Mori, K. & Mori, M. Nitric oxide induced apoptosis in RAW 264.7 macrophages is mediated by endoplasmic reticulum stress pathway involving ATF6 and CHOP. J. Biol. Chem. 277, 12343–12350 (2002).
Toure, B. B. et al. Biosynthesis and enzymatic characterization of human SKI-1/S1P and the processing of its inhibitory prosegment. J. Biol. Chem. 275, 2349–2358 (2000).
Seidah, N. G. et al. Mammalian subtilisin/kexin isozyme SKI-1: A widely expressed proprotein convertase with a unique cleavage specificity and cellular localization. Proc. Natl Acad. Sci. USA 96, 1321–1326 (1999).
Lenz, O., ter Meulen, J., Klenk, H. D., Seidah, N. G. & Garten, W. The Lassa virus glycoprotein precursor GP-C is proteolytically processed by subtilase SKI-1/S1P. Proc. Natl Acad. Sci. USA 98, 12701–12705 (2001).
Mateo, J., Garcia-Lecea, M., Cadenas, S., Hernandez, C., & Moncada, S. Regulation of hypoxia-inducible factor-1α by nitric oxide through mitochondria-dependent and -independent pathways. Biochem. J. 376, 537–544 (2003).
Chen, X., Shen, J. & Prywes, R. The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J. Biol. Chem. 277, 13045–13052 (2002).
Elbashir, S. M. et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411, 494–498 (2001).
Rizzuto, R. et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280, 1763–1766 (1998).
Montero, M. et al. Chromaffin-cell stimulation triggers fast millimolar mitochondrial Ca2+ transients that modulate secretion. Nature Cell Biol. 2, 57–61 (2000).
Nowicky, A. V. & Duchen, M. R. Changes in [Ca2+]i and membrane currents during impaired mitochondrial metabolism in dissociated rat hippocampal neurons. J. Physiol. 507, 131–145 (1998).
Shen, J., Chen, X., Hendershot, L. & Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 3, 99–111 (2002).
Lee, K. et al. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 16, 452–466 (2002).
Drummond, I. A., Lee, A. S., Resendez, E. & Steinhardt, R. A. Depletion of intracellular calcium stores by calcium ionophore A23187 induces the genes for glucose-regulated proteins in hamster fibroblasts. J. Biol. Chem. 262, 12801–12805 (1987).
Beltrán, B., Quintero, M., García-Zaragozá, E., O'Connor, E., Esplugues, J. V. & Moncada, S. Inhibition of mitochondrial respiration by endogenous nitric oxide: a critical step in Fas signaling. Proc. Natl Acad. Sci. USA 99, 8892–8897 (2002).
Acknowledgements
This work was supported by the Medical Research Council, UK. We thank E. A. Higgs for help in the preparation of this manuscript. We also thank R. Prywes for providing the 3×Flag–ATF6 plasmid.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information, Figures
Fig. S1, Fig. S2 and Fig. S3 (PDF 229 kb)
Rights and permissions
About this article
Cite this article
Xu, W., Liu, L., Charles, I. et al. Nitric oxide induces coupling of mitochondrial signalling with the endoplasmic reticulum stress response. Nat Cell Biol 6, 1129–1134 (2004). https://doi.org/10.1038/ncb1188
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ncb1188
This article is cited by
-
Callus induction in Araucaria angustifolia using orthotropic and plagiotropic apexes: proteomic and morphoanatomical aspects
Plant Cell, Tissue and Organ Culture (PCTOC) (2023)
-
Selenium Protects against Lead-induced Apoptosis via Endoplasmic Reticulum Stress in Chicken Kidneys
Biological Trace Element Research (2018)
-
ER Stress and Autophagy in Obesity and Nonalcoholic Fatty Liver Disease
Current Pathobiology Reports (2017)
-
Heat-killed yogurt-containing lactic acid bacteria prevent cytokine-induced barrier disruption in human intestinal Caco-2 cells
Annals of Microbiology (2016)
-
Caveolae and signalling in cancer
Nature Reviews Cancer (2015)
