
Abstract
Upland cotton is a model for polyploid crop domestication and transgenic improvement. Here we sequenced the allotetraploid Gossypium hirsutum L. acc. TM-1 genome by integrating whole-genome shotgun reads, bacterial artificial chromosome (BAC)-end sequences and genotype-by-sequencing genetic maps. We assembled and annotated 32,032 A-subgenome genes and 34,402 D-subgenome genes. Structural rearrangements, gene loss, disrupted genes and sequence divergence were more common in the A subgenome than in the D subgenome, suggesting asymmetric evolution. However, no genome-wide expression dominance was found between the subgenomes. Genomic signatures of selection and domestication are associated with positively selected genes (PSGs) for fiber improvement in the A subgenome and for stress tolerance in the D subgenome. This draft genome sequence provides a resource for engineering superior cotton lines.
Similar content being viewed by others

Main
Allotetraploid Upland cotton (AD)1 (G. hirsutum L.) accounts for more than 90% of cultivated cotton worldwide, is the main source of renewable textile fibers and is also grown to produce oilseed1. Cotton was among the earliest adopted genetically modified crops, and more than 70% of cotton crops are transgenic (http://www.isaaa.org/resources). The biotechnological improvement of cotton could be further accelerated by decoding the allotetraploid genome1. Although the genomes of two extant progenitor relatives G. arboreum2 and G. raimondii3,4 have been sequenced, the exact donor species that led to the formation of the tetraploid cotton species 1–2 million years ago (MYA) no longer exists5 (Fig. 1). Therefore, to fully understand the evolution of G. hirsutum and gain insights into cotton fiber biology, it is necessary to sequence the allotetraploid species.

Fiber phenotypes of domesticated allotetraploid cotton (G. hirsutum L. acc. TM-1), wild cotton (G. hirsutum L. acc. yucatanense), and their two closest extant progenitors, G. herbaceum and G. raimondii. Scale bar, 10 mm.
One of the challenges in sequencing polyploid genomes is the difficulty in discriminating between homoeologous sequences. In species with large chromosomes, such as wheat, individual chromosomes or arms can be sorted before sequencing6. However, chromosome sorting is less useful for cotton, which has a large number of small chromosomes (2n = 4x = 52). A conventional approach is to sequence diploid progenitor genomes, which can then guide the assembly of homoeologous chromosomes of allopolyploids, as was done for Brassica napus7,8,9. One problem with this approach is that many sequence contigs and scaffolds remain ambiguous with respect to homoeologous relationships.
Here we developed an integrated approach to sequence and assemble the genome of the allotetraploid cotton G. hirsutum L. acc. Texas Marker-1 (TM-1), which is widely used as a genetic standard1,10. We generated a genome sequence containing two- to about fourfold longer sequence scaffolds (N50 = 1,600 kb) than that produced for other allopolyploid species Brassica napus (N50 = 764 kb)9, Nicotiana tabacum (N50 = 345∼386 kb)11 and wheat (contig N50 = 515∼4,297 bp)6, as well as the extant diploid A-genome progenitor of G. arboreum (N50 = 666 kb)2 (N50, the size above which 50% of the total length of the sequence assembly can be found). Compared with canola and wheat, which emerged 4,000–8,000 years ago, allotetraploid cotton is much older (1–2 MYA), and its subgenomes have undergone asymmetric evolution, including biased subgenomic arrangements and transposable element amplification, selective gene loss, unequal distribution of disrupted genes, and expression bias of homoeologous genes related to fiber development and wider adaptation, respectively.
Results
Assembly of the G. hirsutum genome
An allohaploid plant was derived from the allotetraploid cotton (TM-1) and used for genome sequencing. We produced 612 Gb (245× genome equivalent) of high-quality Illumina reads, which were assembled using SOAPdenovo12 (Supplementary Table 1). The resulting contigs and scaffold were integrated using 174,454 pairs of Sanger-sequenced BAC-end sequences comprising 116.5 Mb, and assembled into the TM-1 genome sequence (V1.0). To correct for misassembly, classify the homoeologous segments and order the scaffolds, we developed an ultradense genetic map using genotyping by sequencing of 59 F2 individuals derived from TM-1 and G. barbadense cv. Hai7124. The map consisted of 4,999,048 single-nucleotide polymorphism (SNP) loci and 4,049 recombination bins spanning 4,042 cM in 26 linkage groups. Using the map, we corrected 218 misassembled scaffolds (442.2 Mb, or 17.6%, of the genome sequence) in the assembly V1.0 and found that most misassembled scaffolds were caused by ambiguous homeolog sequences (Supplementary Table 2). The final assembly (V1.1) comprised 265,279 contigs (N50 = 34.0 kb) and 40,407 scaffolds (N50 = 1.6 Mb) (Supplementary Table 3). The total scaffold length (2.4 Gb) spanned ∼96% of the estimated allotetraploid genome (2.5 Gb) (Supplementary Fig. 1), of which 6,146 scaffolds (2.3 Gb) were aligned and organized into 26 pseudochromosomes, including 1.5 Gb (4,635 scaffolds) in the A subgenome and 0.8 Gb (1,511 scaffolds) in the D subgenome. Furthermore, 1.9 Gb (79.2%) was oriented based on linkage maps (Supplementary Table 3).
Validation of the assembly
More than 97.7% of the genome was covered by more than tenfold paired-end sequence reads (Supplementary Fig. 2). The genome assembly accuracy and completeness were validated by matching 98% of mRNA sequences in G. raimondii4 (Supplementary Table 4) and 99.5% of full-length mRNAs of G. hirsutum, which were downloaded from the NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank/) (Supplementary Table 5). This indicates that the sequence contained most genic regions. The sequence showed near-perfect matches with 36 completely sequenced BACs13,14 (Supplementary Fig. 3 and Supplementary Table 6) and consistency with the integrated genetic and cytogenetic maps of A12 and D12 homoeologous chromosomes using fluorescence in situ hybridization (FISH) (Supplementary Fig. 4a)15, further confirming the assembly quality of the allotetraploid genome sequence.
Genome annotation
The allotetraploid cotton sequence consists of 70,478 predicted protein-coding genes with an average length of 1,179 bp and five exons per gene (Supplementary Table 7). Of these, 67,736 (96.1%), including 32,032 in the A subgenome and 34,402 in the D subgenome (Supplementary Fig. 5 and Supplementary Table 3), were functionally annotated (Supplementary Table 8). The protein-coding genes are unevenly distributed across each chromosome with the gene density increasing toward the chromosomal ends (Fig. 2a). There are 4,778 putative transcription factors from 59 families16, representing 6.8% of the protein-coding genes (Supplementary Table 9). In addition, homology-based annotation of noncoding RNA sequences predicted 2,226 transfer RNA genes, 301 microRNA genes, 885 small nuclear RNA genes and 1,061 ribosomal RNA genes (Supplementary Table 10).

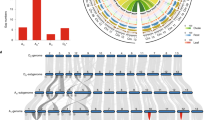
(a) Allotetraploid cotton (TM-1) genome topography and synteny. I indicates the oriented homoeologous sites of duplicated gene blocks within TM-1. II represents the frequency distribution of recombination nodules (RNs), MAX = 25. III represents the genomic overview of 26 assembled TM-1 chromosomes composed of exons (red), introns (blue), Copia retrotransposons (yellow) and Gypsy retrotransposons (purple). IV indicates the chromosome karyotype. Colored segments correspond to the A and D homoeologous chromosomes. (b) Distribution of Ks values for 21,618 one-to-one orthologous gene sets between the A genome2, D genome4 and two subgenomes of TM-1. AtDt; 't' indicates tetraploid. Peak values for each comparison are indicated with arrows. (c) Comparison of Ka and Ks distributions in the A and D subgenomes. The central line for each box plot indicated median. The top and bottom edges of the box indicated the 25th and 75th percentiles and the whiskers extend 1.5 times of the interquartile range beyond the edges of the box. The asterisks represent significant differences (Wilcoxon rank sum test, P < 0.05). (d) Genome composition of the A genome2, D genome4 and two subgenomes of TM-1. (e) Comparison of biased expressed genes, PSGs, loss genes and disrupted genes in the two subgenomes. *P < 0.01, Fisher's exact tests.
Comparative genome analyses
Polyploidization results in whole-genome duplication, which is often followed by a diploidization process that is characterized by rapid genomic reorganization and massive gene loss17,18,19. This process has been observed in paleopolyploid maize20 and in newly synthesized allopolyploid wheat21, Brassica22 and Tragopogon23. Some other allopolyploids, such as cotton24 and Arabidopsis25, do not show many changes in their genomic sequences. Recent genome sequencing studies revealed a relatively stable genome organization in cultivated hexaploid wheat6 and Brassica napus9. This is probably because the exact progenitors that could form stable cultivated allopolyploids were lost. Alternatively, unstable allotetraploids in early generations were eliminated by selection. Here we show that in allotetraploid cotton, overall gene order and colinearity are largely conserved between the A and D subgenomes and the extant D-progenitor genome (G. raimondii)4 (Fig. 2a, Supplementary Figs. 6a and 7). However, this colinearity was not obvious with either the A-progenitor genome (G. arboreum)2 or another version of the D-progenitor genome (G. raimondii)3 because of numerous mis-assemblies. For example, many scaffolds from several different pseudochromosomes were present in one pseudomolecule (Supplementary Figs. 4b and 6b,c). Therefore, we used the G. raimondii genome4 for further comparative genomic analysis. In the G. hirsutum draft genome, at least nine translocations and 28 inversions were identified (Supplementary Fig. 8, Supplementary Tables 11 and 12), including two large reciprocal translocations between A02 and A03 and between A04 and A05 (Fig. 2a, Supplementary Figs. 6 and 8), that have also been confirmed by cytogenetic data26. In addition, three inversions on the A12 and D12 homoeologous chromosomes compared to Gr_Chr08 (ref. 4) were validated by BAC-FISH15 (Supplementary Fig. 4). Overall, the number of rearrangements between the A and D subgenomes (19 versus 18) was similar. However, the length of total rearrangements was larger in the A subgenome (372.6 Mb) than in the D subgenome (82.6 Mb) (Supplementary Tables 11 and 12). The average length per rearrangement was 19.6 Mb in the A subgenome, which is significantly larger than that (4.6 Mb) in the D subgenome (t-test, P = 0.0064).
Asymmetric evolution of the A and D subgenomes
By comparing the synonymous substitution values (Ks) of 21,618 orthologous gene sets between the sequenced progenitor genomes2,4 and the two subgenomes, the divergence time between the A- and D-progenitor genomes was estimated to be 6.0–6.3 MYA (Ks peaks at 0.031 and 0.033, respectively), and the allotetraploid was formed approximately 1–1.5 MYA (Ks peaks at 0.005 and 0.008, respectively) (Fig. 2b). Based on the phylogenetic tree constructed using 6,911 orthologous gene families from Theobroma cacao27, G. raimondii4, G. arboreum2 and the two subgenomes of G. hirsutum, the evolution rates of homologous genes were estimated for each branch (Supplementary Fig. 9a). Both the nonsynonymous substitution rate (Ka) and Ks values were elevated in the A and D subgenomes compared with their corresponding progenitor genomes, indicating faster evolution rates in allotetraploid cotton (Supplementary Fig. 9b,c). Interestingly, the A subgenome might have evolved faster than the D subgenome, suggesting asymmetric evolution of the two subgenomes (Fig. 2c). Asymmetric evolution could be caused by relaxed selection pressure in the A subgenome (Supplementary Fig. 9d). Tajima's relative rate test for the evolutionary rate confirmed this result (Supplementary Table 13).
The assembled A subgenome (1,477 Mb) is nearly twice the size of the D subgenome (831 Mb), which is consistent with the genome size difference between G. arboreum2 and G. raimondii3,4. At least 64.8% of the assembled genome are transposable elements (TEs), which was substantially higher than that in canola (>34.8%)9, but lower than in wheat (>76.6%)6. Although the total content of TEs was similar to that present in a second G. hirsutum draft genome28 (64.8% versus 66%), the retrotransposon frequencies were different (52.29% versus 62.81%). This variance was most likely caused by the different stringency applied for TE classification by the reference databases. We used the database of the Munich Information Center for Protein Sequences (MIPS), which incorporates all the TEs identified from the D-progenitor genome4, including those identified manually so it is possible that this database provides a relatively accurate estimation of retrotransposon numbers and types. It might, however, miss some uncommon elements, most of which were defined as unclassified elements in our data set. The Repbase and ab initio databases were used for retrotransposon identification in a second G. hirsutum paper28, which might include additional TEs or other elements that were not present either in MIPS or in our analysis. Our analysis shows that there were more TEs in the A subgenome (at least 843.5 Mb) than in the D subgenome (at least 433 Mb). Among them, the number of Gypsy retroelements (25.33%, 523.85 Mb) was threefold higher in the A subgenome (362 Mb) than in the D subgenome (136 Mb) (Fig. 2d and Supplementary Table 14). The TE types and relative proportions were similar between the A subgenome and A-progenitor (A2) genome2 and between the D subgenome and D-progenitor (D5) genome4 (Supplementary Fig. 10 and Supplementary Table 14). The distribution of TE divergence levels (Kimura distance > 0.01, older than 1.5 MYA) suggests that most TEs expanded before the formation of allopolyploid cotton (Supplementary Fig. 10). It is likely that the TE expansion occurred in the progenitor genomes and was retained after allopolyploid formation.
Gene loss during cotton evolution
Gene loss in the allotetraploid cotton genome is rare; 228 genes were lost in the A subgenome and 141 in the D subgenome (P < 0.01, Fisher's exact test; Fig. 2e and Supplementary Table 15). In addition, we identified 4,312 disrupted genes, which are defined as those genes that contain frame shift or premature stop codons compared to their orthologous genes29. Consistent with asymmetric gene loss, there were significantly more disrupted genes in the A subgenome (2,425) than in the D subgenome (1,887) (P < 0.01, Fisher's exact test, Fig. 2e). The average expression levels of these disrupted genes were significantly lower than that of all other genes in their corresponding subgenome (P < 0.01, Wilcoxon rank sum test; Supplementary Fig. 11). This suggests that gene loss is most likely an ongoing process in allotetraploid cotton.
Expression of homoeologous genes in allotetraploid cotton
Another consequence of polyploidization is unequal expression of homoeologous genes in allopolyploids30,31,32, but whole-genome analyses of homoeologous gene expression have gotten inconsistent results4,33. In our study, ∼90% of 25,358 homoeologous gene pairs were expressed (fragments per kilobase of transcript per million reads sequenced (FPKM) > 1) in 35 vegetative and reproductive tissues (Supplementary Table 16). Although the average expression levels and numbers of expressed genes varied among different tissues and developmental stages (Supplementary Fig. 12), homoeologous gene pairs did not show genome-wide expression bias (Supplementary Fig. 13). However, in a specific tissue or developmental stage, 20–40% of homoeologous gene pairs showed biased A- or D-homoeolog expression (Supplementary Table 17), which is consistent with the published data4,34 and may lead to subfunctionalization because of expression divergence35. There were slightly more genes with expression bias toward the D homoeologs than toward the A homoeologs (Supplementary Fig. 14). Although the number of genes that were biased toward either A- or D-homeolog expression in cotton fiber elongation (10 days post-anthesis (DPA)) and secondary cell wall synthesis (20 DPA) were similar to those previously reported34, more transcription factor genes (such as MYB family members) were expressed in the A homoeologs (Supplementary Fig. 15), suggesting important roles in fiber development36.
Cotton fiber biology and domestication
MYB-domain transcription factors are important for cotton fiber and leaf trichome development36,37,38. Among the ten MYB MIXTA-like (GhMML) homoeologs in allotetraploid cotton (Supplementary Fig. 16), all were highly expressed during fiber initiation in cultivated cotton (Fig. 3a,b). One group of GhMML genes was downregulated in three natural fiberless mutants (Fig. 3c,d), which resembled the fiberless seeds produced in GhMYB25-like RNA interference–suppressing lines37, but trichomes developed normally elsewhere. They are different from the GL1-like GhMYB2 (ref. 38) and GhMYB109 groups, which primarily promote leaf trichome development in cotton. These two groups of genes in cotton are predicted to affect leaf trichome and seed fiber development, respectively.

(a) Scanning electron microscope (SEM) image of 0 DPA ovule epidermis (scale bar, 20 μm) and photograph of a mature boll of wild type (scale bar, 1 cm). (b) Heat map of the expression of 20 MYBMIXTA-like genes in 13 tissues. We sampled fiber-bearing ovules on −3, −1, 0, 1 and 3 DPA; 5, 10, 15 and 20 DPA are fiber samples. (c) SEM images of 0 DPA ovule epidermis (scale bar, 20 μm) and photographs of a boll of fiberless mutant (scale bar, 1 cm). (d) Heat map of the expression of 20 MYBMIXTA-like genes in the fiberless mutants, SL1-7-1, MD17 and Xuzhou142 (XZ142), compared to wild-type TM-1(W).
Compared to the progenitor species G. arboreum and G. raimondii, Upland cotton has not only developed longer, stronger and finer fibers, but has also adapted to more diverse environments. However, the genomic basis of this is largely unknown. The genomic signatures of selection and domestication comprised 811 PSGs39, including 470 and 341 in the A and D subgenomes, respectively (Fig. 2e and Supplementary Table 18). PSGs refer to those genes with the codons subjected to positive selection. Among this set, 591 PSGs (72.8%) were expressed during fiber development, which probably results from the domestication for cotton fiber production. Interestingly, these A- and D-subgenome PSGs were enriched in different gene ontology groups. Many of the A-homoeologous PSGs (343/470) were enriched in the synthesis of ethylene and very-long-chain fatty acids, which are known to affect fiber elongation40, and in sucrose metabolism and beta-D-glucan biosynthetic pathway to produce UDP-glucose, the preferred substrate for cellulose biosynthesis34,41,42 (Fig. 4a). The D-homoeologous PSGs (248/341) were enriched in carbohydrate transport, response to superoxide and other abiotic stresses (Supplementary Table 19). These results suggest that allotetraploid cotton domestication is associated with intensive human selection for fiber yield and quality on the A homoeologs from fiber-producing species and for wider adaptation on the D homoeologs from non-poor species.

(a) Pathways of carbohydrate and fatty acid metabolism related to cell wall biosynthesis. Blue type indicates the gene family associated with domestication. *, genes that are related to QTL hotspots for fiber length and strength. The full names of these gene families are shown in Supplementary Table 22. (b) Phylogenetic relationships of G. hirsutum CESAs. These CESAs were classified into two groups, primary cell wall (PCW) in orange lines and secondary cell wall (SCW) in blue lines. (c) Heat map of CESAs expression patterns in 22 cotton tissues. (d) Expression levels of SCW-related CESAs in domesticated cotton (TM-1) and two wild relatives (TX665, G. hirsutum var. palmeri; TX2094, G. hirsutum var. yucatanese). Fisher's exact test, *P value < 0.05 and fold-change > 2.
Consistent with the above notion, 70 PSGs in the A subgenome that are related to fiber development, including phosphoenolpyruvate carboxylase gene (PEPC)43, are upregulated in domesticated cotton, compared to wild relatives of G. hirsutum race palmeri and yucatanense4. In the D subgenome, 54 upregulated PSGs (Supplementary Table 20) are related to salt and/or osmotic stress tolerance, response to superoxide and ethylene, and include radical-induced cell death 1 gene (RCD1)44 (Gh_D08G1685) and aldehyde dehydrogenase 7 (ALDH7)45 (Gh_D06G1578) (Supplementary Fig. 17).
Genomic selection for superior fiber quality was further supported by the analysis of 952 quantitative trait loci (QTLs) in allotetraploid cotton. Forty-one PSGs are located within seven QTL hotspots associated with fiber length and strength (Supplementary Table 21). These PSGs in the A subgenome include genes encoding peroxidase superfamily proteins (POX)40 and cellulose synthases41, which are known to affect cell wall biosynthesis (Fig. 4a and Supplementary Table 22). PSGs in the D subgenome such as RCD1 and NAC transcription factor genes46 are predicted to facilitate fiber development by regulating abiotic stress tolerance. These data suggest that fiber-related genes in the A subgenome contribute to fiber improvement, which is consistent with the phenotype in the A progenitor but not in the D progenitor. Previous studies on fiber-related QTLs within the D subgenome could be related to trans-acting and/or epistatic effects1.
Among the D-subgenome PSGs that are enriched in GO groups including responses to water, salt, superoxide and other abiotic stresses, 68 were upregulated in response to four different stress treatments in Upland cotton (Supplementary Tables 23 and 24). This indicates that the D subgenome contributes to stress tolerance during allotetraploid cotton domestication. Thus, A and D subgenomes contribute to fiber and adaptation traits, respectively.
Mature cotton consists of >90% crystalline cellulose with cellulose synthase (CESA) required for cellulose biosynthesis during secondary-wall thickening41. There are at least 32 CESA family genes in allotetraploid cotton, which is twice as many as in G. raimondii4 (Supplementary Table 25). These CESA family members can be classified into two major groups with six branches (Fig. 4b). One group was expressed during primary cell wall development and in many other tissues (Fig. 4c). The other group including CESA4, CESA7 and CESA8 was predominately expressed during secondary cell wall biosynthesis (Fig. 4c) and in 20–25 DPA at levels 1.5- to 40-fold higher in TM-1 than in wild cottons (Fig. 4d), suggesting their potential role in increased lint yield and improved qualities in domesticated cotton. Together, these new genomic data provide guidelines to improve agronomic traits in allotetraploid cotton.
Discussion
Although new sequencing technologies have dramatically reduced costs, assembling short reads remains a challenge for complex polyploid genomes. Polyploidy generates genetic and gene expression novelty but with this comes redundancy that impedes sequence annotation and assembly. We have developed an integrated approach to the sequencing and assembly of an allopolyploid cotton genome that could be applied to the sequencing of complex genomes of other polyploid crops. In contrast to recently formed allopolyploids such as wheat6 and canola9, the allopolyploid cotton genome has features of asymmetrical evolution. This sequence of an allotetraploid cotton line, together with genetically mapped QTLs and ∼5 million SNPs between G. hirsutum and G. barbadense, provide genomic resources for map-based cloning, germplasm improvement and genomic selection in cotton lines to help meet the growing demand for renewable fiber, oil and fuel.
Methods
Production of an allohaploid plant of the standard genetic line, TM-1, for sequencing.
An allohaploid plant of Texas Marker-1 (TM-1), G. hirsutum, (AD)1 was isolated by screening ∼2,400 F1 individuals from the cross (Vsg × TM-1). Vsg is a virescently marked semigametic line of G. barbadense (AD)2 (ref. 47). The resulting plant consists of allohaploid-hirsutum and allohaploid-barbadense, which can be visually discriminated by the Vsg phenotypic marker. In addition, the paternal allohaploid plants were cytogenetically confirmed in pollen mother cells at meiosis, and further validated by whole-genome genotyping with 402 microsatellite pairs selected at 10-cM intervals from our linkage map13.
Genome sequencing data generation.
DNA from the allohaploid TM-1 plant was prepared using the CTAB extraction method. The genomic DNA was sheared with a Bioruptor sonication device (Diagenode SA., Liege, Belgium) for short-insert paired-end (PE) library construction, and with a Hydroshear DNA Shearing Device (Genomic Solutions Inc., Ann Arbor, MI, USA) for mate-pair library construction. Short-insert paired-end (180, 300, and 500 bp) and large-insert mate-pair libraries (2, 5, 10 kb) were prepared according to the manufacturer's instructions (Illumina, San Diego, CA). All libraries were sequenced at 2 × 100 bp on an Illumina HiSeq 2000 platform. In total, 843 Gb of DNA sequencing read data were generated for the genome assembly, representing approximately 337-fold coverage of the raw genome.
BAC library construction and BAC end sequencing.
Three BAC libraries (GH_TBh, derived from partial restriction digestion with HindIII; GH_TBb, derived from partial restriction digestion with BstYI; and GH_TBr, derived from random shearing) with average insert sizes of 160 kb, 152 kb and 100 kb, respectively, were sequenced at both ends using the Sanger sequencing method. Each sequence was trimmed to remove BAC vector sequences and low-quality bases (<phred20). A total of 179,209 BAC-end sequences with an average size of 400 bp (116 Mb) were derived from the three independent BAC libraries. After filtering the single BAC-end sequences, we obtained 174,454 paired BAC-end sequences (42,643 BAC-ends from the HindIII library, 40,672 BAC-ends from the BstYI library and 3,912 BAC-ends from the randomly sheared library) that were aligned to the temporary scaffolds using the long sequence alignment module of Burrows-Wheeler Aligner. Approximately 96% of the BAC-end sequences with >90% of full length were mapped to the temporary scaffolds.
Genome size estimation.
The distribution of K-mer frequency, also known as kmer spectrum, is widely used for the estimation of genome size. We adopted a method based on K-mer distribution to estimate the genome size with 72× high-quality reads (181 Gb) from short-insert size libraries (≤800 bp). We obtained a 31-nucleotide depth distribution and observed that the peak depth was at 70. The following formula48 was applied: genome size = total_kmer_num/kmer_depth, where total_kmer_num is the total number of K-mers in the sequence, and kmer_depth is the peak depth on the K-mer frequency distribution map. Using this method, the size of the G. hirsutum genome is estimated to be 2.5 Gb.
Genome assembly, scaffolding and gap-closing.
All sequences were assembled using the SOAPdenovo package12. A de Bruijn graph was built using a K-mer size of 63. After removing tips, merging bubbles and concatenating the tiny repeats, contigs were built from the simplified de Bruijn graph. Paired-end short reads were then aligned back onto the contigs to construct the linkage relationship for contigs. Scaffolds were assembled based on these paired-end links and gaps in the scaffolds were filled by Gapcloser. BAC-ends were mapped to the assembly using BWA-SW software12. Further scaffolding was then conducted, based on links between BAC-ends.
Linkage map construction using the TM-1 × Hai7124 mapping population.
An ultra-high-density genetic map was created using an F2 population of a cross of TM-1 × Hai7124. DNA libraries of TM-1, Hai7124 and 59 of the 249 F2 individuals were sequenced (2× 100-bp PE reads) on the Illumina HiSeq 2000 platform. The sequencing depth was 39.1-fold for Hai7124 and averaged 5.3-fold for the 59 F2 individuals. 5,054,468 simple SNPs between TM-1 and Hai7124 were identified using Samtools49. After filtering out the low-coverage SNPs and genotyping the sequenced F2 individuals, an ultra-high-density genetic map (4,999,048 simple SNPs) was constructed using JoinMap3.0 and MapChart software50,51.
Correction of the TM-1 assembly using the SNP map and pseudomolecule chromosome construction.
The ultradense genetic linkage map and the synteny with G. raimondii were used to identify false joins and to integrate the assembled scaffolds into 26 pseudomolecule chromosomes. Scaffolds were broken if they contained a putative false join based on the genetic map. The corrected scaffolds were then concatenated to create 26 chromosomes based on multiple markers, synteny with G. raimondii and positive relationships of PE reads. A total of 1,430 joins were made on 1,456 scaffolds to form the final assembly containing 26 chromosomes capturing 1.9 Gb (79.2%) of the assembled sequences. Each join was padded with 10,000 Ns.
Assessment of genome assembly quality by PE reads.
To evaluate the quality of the genome assembly, we first mapped the reads from short-insert-size libraries (180 bp) back to the scaffolds using BWA12 with the following parameters: -o 1 -i 15. The sequencing depth distribution followed a Poisson distribution, which indicates the uniformity of the genome sequencing process. A total of 98.60% of the reads could be mapped back to the current assembly, which covered over 99.64% of the assembly, excluding gaps. This observation implies that the current assembly covered almost all unique genome regions. About 94.7% of the assembly was covered by at least 20× reads, which guaranteed the high level of accuracy of the assembly at the single-nucleotide level. The scatter graph of the distribution of GC content versus sequencing depth was plotted and only one major depth island was present on the distribution graph. This suggests that there was no major contamination from other sources such as bacteria or chloroplasts that could severely affect the current genome assembly.
TM-1 assembly validation using mRNA sequences from the G. raimondii and G. hirsutum genomes.
To evaluate the completeness of the genome assembly in genic regions, we used whole mRNA sequences from the G. raimondii genome4 as queries in BLAST searches against the TM-1 assembly with default parameters. Of the 37,505 G. raimondii genes, 99.82% were mapped to the TM-1 assembly, and 98.24% were considered as complete sequences on the assembly (more than 90% of the mRNA could be mapped to one continuous scaffold). Owing to the allopolyploid nature of the TM-1 genome, the D-genome (represented by G. raimondii) represents one of two subgenomes in the tetraploid. Thus, the full-length mRNA sequences from G. hirsutum were downloaded from GenBank. Of the 1,054 full-length mRNAs, 99.5% were mapped to the TM-1 assembly, and 95.1% were considered as complete sequences on the assembly; higher values than those from previous G. raimondii genome sequencing (90.3%)4.
TM-1 assembly validation using 36 completely sequenced BACs.
To examine the accuracy of the assembly, we downloaded 36 full-length BAC clone sequences from GenBank; 32 from G. hirsutum cv. Maxxa (http://www.ncbi.nlm.nih.gov/nuccore), and 4 from TM-1 (ref. 13,14). These BAC clones were aligned to the TM-1 assembly using LASTZ software52. All clones were almost intact in our assembly (>98% of their lengths were covered by only one scaffold).
Gene prediction and annotation.
Protein-coding region identification and gene prediction were conducted through a combination of homology-based prediction, de novo prediction, and transcriptome-based prediction methods. Proteins from seven plant genomes (Arabidopsis thaliana, Carica papaya, Glycine max, G. raimondii, Populus trichocarpa, Theobroma cacao and Vitis vinifera) were downloaded from Phytozome (Release 9.1)53. Protein sequences were aligned to the assembly using TblastN54 with an E-value cutoff of 1e-5. The BLAST hits were conjoined by Solar software55. GeneWise56 was used to predict the exact gene structure of the corresponding genomic regions on each BLAST hit. Five ab initio gene prediction programs, Augustus (version 2.5.5)57, Genscan (version 1.0)58, GlimmerHMM (version 3.0.1)59, Geneid60, and SNAP61, were used to predict coding regions in the repeat-masked genome. Finally, RNA-seq data were mapped to the assembly using Tophat (version 2.0.8)62. Cufflinks (version 2.1.1)63 (http://cufflinks.cbcb.umd.edu/) was then used to assemble the transcripts into gene models. All gene models predicted from the above three approaches were combined by EvidenceModeler (EVM) into a nonredundant set of gene structures64. The gene models were further filtered based on their Cscore, peptide coverage and its CDS overlapping with TEs. Only transcripts with a C-score ≥ 0.5, and peptide coverage ≥ 0.5, were retained. Finally, gene models with more than 30% of their coding peptides annotated as Pfam65 or Interprot66 TE domains were filtered out. Functional annotation of protein-coding genes was achieved using BLASTP67 (E-value 1e-05) against two integrated protein sequence databases; SwissProt and TrEMBL68. Protein domains were annotated by searching against the InterPro (V32.0)66 and Pfam (V27.0) databases65, using InterProScan (V4.8) and HMMER69 (V3.1), respectively. The Gene Ontology70 (GO) terms for each gene were obtained from the corresponding InterPro or Pfam entry. The pathways in which the genes might be involved were assigned by BLAST against the KEGG databases (release 53)71, with an E-value cutoff of 1e-05.
Transcription factors annotation.
The Hidden Markov Model (HMM) profiles of gene domains were downloaded from Pfam (http://pfam.sanger.ac.uk/) for use in gene family identification. HMMER3.0 (ref. 69) and BLASTP67 searches were used to identify putative genes in the TM-1 genome. The candidate genes were confirmed in the Pfam database65. Transcription factors were classified into 59 families based on the default parameters for transcription factor domain structure from PlnTFDB version 3.0 (ref. 16).
Noncoding RNAs annotation.
Noncoding RNA genes, including rRNA genes, tRNA genes and snRNA genes were predicted in the assembly. The tRNA genes were identified by tRNAscan-SE72 software with the eukaryote parameters. The rRNA fragments were predicted by aligning to Arabidopsis and rice template rRNA sequences using BlastN at an E-value of 1e-10. The miRNA and snRNA genes were predicted by searching against the Rfam database (release 9.1)73 using INFERNAL software74.
TE annotation.
TEs in the TM-1 genome were identified by RepeatMasker (version 3.3.0) (http://www.repeatmasker.org/) against the Malvid section of the MIPS repeat database (mipsREdat_9.3p)75. RepeatMasker results were filtered to retain the high-confidence hits (length ≥ 50 bp, score ≥ 255). To estimate the potential age of TE types, we calculated Kimura distances between TE copies (consensus from the TE library versus copy in the genome). The Kimura distance was estimated through combination of transition rate and transversion rate, using the formula (K = −(1/2) ln(1−2p−q)−(1/4) ln(1−2q)). 'p' is the proportion of transition site and 'q' is the proportion of transversion site. Using Kimura distances, relative age of different TE types in the genome was plotted.
Identification of homoeologous gene sets and orthologous gene sets.
An OrthoMCL clustering program was run on the proteome from the G. raimondii4, Theobroma cacao and G. arboreum2 genomes, and the two subgenomes of G. hirsutum, as downloaded from Phytozome53. One-to-one orthologous gene sets were then extracted from the clustering results. A total of 6,911 one-to-one orthologous gene sets were found between T. cacao, G. raimondii4, G. arboreum2 and the two subgenomes of G. hirsutum. In addition, a total of 21,618 one-to-one orthologous gene sets were found between G. raimondii4, G. arboreum2 and the two subgenomes of G. hirsutum.
Estimation of divergence time.
Based on the 21,618 cotton orthologous gene sets between G. raimondii4, G. arboreum2 and the two subgenomes of G. hirsutum, the synonymous divergence levels (Ks) between all three cotton species were calculated. The formula “t = Ks/2r” was used to estimate the divergence time between two subgenomes, where “r” is the neutral substitution rate. A neutral substitution rate of 2.6 × 10−9 was used in the current study76.
Phylogenetic tree construction and evolution rate estimation.
A phylogenetic tree was constructed for the five genomes (T. cacao, G. raimondii4, G. arboreum2 and the two subgenomes of G. hirsutum) using the coding sequence (CDS) alignment of the 6,911 orthologs of these genomes. Evolutionary rates (Ka and Ks) on each branch were estimated using the codeml program in the PAML package77. We used the free-ratio “branch” model, which allows distinct evolutionary rates for each branch. Significant rate variations between linages were examined through Wilcoxon rank sum tests. In addition, a Tajima's relative rate test (RRT), as implemented in the MEGA6 package78, was used to validate the results. Each comparison in RRT included two ingroups and one outgroup. All tests were done using the T. cacao as outgroup.
Syntenic analysis and whole-genome alignment.
Homology searches were carried out using BLASTP (E-value threshold 1e-5). Syntenic blocks were required to match two criteria: at least 30 genes per block in the T .cacao, G. raimondii4 and TM-1 genomes, and at least five genes per block between the subgenomes and the D-genome. The criteria tests were conducted with MCscan. After the repeat regions were masked, whole-genome alignment of TM-1 and G. raimondii genomic sequences, as reported by both the Joint Genome Institute (JGI) and BGI-Shenzhen, was carried out using the MUMmer 3 package (v. 3.23) in order to study the intergenomic relationships between these species.
PSGs.
Using the phylogenetic tree described in (Supplementary Fig. 9a), the branch-site model79 incorporated in the PAML package was used to detect PSGs. To detect PSGs in the A subgenome, the branch of the A subgenome was used as the foreground branch and all other branches in the phylogeny were used as background branches. Similar approaches were used to detect genes that were positively selected in the D subgenome. The null model used in the branch-site test assumed that the Ka/Ks values for all codons in all branches must be ≤1, whereas the alternative model assumed that the foreground branch included codons evolving at Ka/Ks >1. A maximum likelihood ratio test (LRT) was used to compare the two models. The p value was calculated through the chi-square distribution with 1 degree of freedom (df = 1). Then the p values were adjusted for multiple testing using the false discovery rate (FDR) method. Genes were identified as positively selected when the FDR < 0.05. Furthermore, we required that at least one amino acid site possessed a high probability of being positive selected (Bayes probability >95%). If none of the amino acids passed this cutoff in the PSGs, these genes were identified as a false positive and excluded.
Gene loss.
Putative gene-loss events were traced from the synteny table using the flanking gene method. Given flanking genes A, B and C in order, if gene B is present in two progenitor genomes and one of the subgenomes, but missed in the other subgenome, then gene B is denoted as a possible lost gene in the allotetraploid. Due to the fragmented nature of the genome assembly, both A and C are required to be present in all four cotton genomes. To avoid false positives due to the failure of gene annotation or accelerated evolution of gene sequences, the intergenic sequence between genes “A” and “C” was extracted in this study. A GeneWise prediction in this intergenic region was then carried out using the B protein sequence. If the predicted protein could be aligned to the original protein with identity >40% and coverage >20%, this gene loss event was defined as a false positive and filtered out.
Genes involved in the ongoing process of gene loss.
Given that allotetraploidization occurred less than 2 MYA, in addition to genes defined as lost, more genes are probably in the process of becoming lost from the genome. These genes were identified through BLAST alignment and GeneWise prediction. Protein-coding sequences of the D genome were BLASTed against the A and D subgenomes. Only ortholog alignments were retained. Then the best-matched location of each gene was predicted for gene structure using GeneWise. Genes containing premature stop codons or frame shift mutations were identified. These genes were defined as disrupted genes that might be involved in the ongoing process of gene loss.
Samples for RNA-seq.
To obtain a comprehensive profile of the TM-1 transcriptome, we used the Illumina Hiseq2000 to perform high-throughput RNA-seq of 56 plant tissue samples: leaves, roots and stems of 2-week-old plants; petals, torus, pistils, stamens and lower sepals dissected from whole mature flowers; ovules from −3, −1, 0, 1, 3, 5, 10, 20 and 25 days DPA; fibers from 5, 10, 20 and 25 DPA; true leaves of the seedlings treated with salt, PEG, heat and cold. In total, 297.3 Gb of raw RNA-seq data were generated from the 57 libraries. In addition, the raw fiber transcriptome data of three wild cottons including G. hirsutum var. palmeri (TX665) and G. hirsutum var. yuncatanense (TX2094 and TX2095) were downloaded from SRX202873 (ref. 4).
Calculation of gene expression levels.
RNA-seq reads were mapped to the TM-1 genome using Tophat62 (Version 2.0.8). To measure the gene expression level in 57 tissues at different sequencing depths, we calculated the expression of each gene using FPKM (Fragments per Kilobase of exon model per Million mapped reads) with Cufflinks (Version 2.1.1)63 (http://cufflinks.cbcb.umd.edu/).
Expression bias of homoeologs.
Protein coding genes from the A and D subgenomes were used as queries in BLAST searches against one another. Gene pairs that displayed the best reciprocal BLAST hits between the two subgenomes were extracted. Thus, 25,358 homoeologous gene pairs were obtained. To analyze the global expression pattern of the two subgenomes, we compared the average FPKM values of 25,358 homoeologs and the expressed tissue numbers among 35 tissues (root, stem, leaf, petal, torus, pistil, stamen, sepal, calycle, 13 tissues from seed germination, ovules at ten different developmental stages, and fibers from 5, 10, 20, and 25 DPA). Homoeologous bias expression was detected in the entire 35 tissue data set through pairwise t-tests (significance was set at P < 0.01, FDR < 0.05 and at least twofold differences in average expression levels). At > Dt indicated biased expression of the A homoeologs and Dt > At indicated biased expression of the D homoeologs. To evaluate expression bias between homoeologous gene pairs in each tissue, homeolog-specific read counts for each gene pair were compared using Fisher's exact test. The P values were adjusted using the BH method79. Then differential expression between gene pairs was delimited by 1.5-fold expression changes and with an FDR < 0.05.
Analysis of differentially expressed genes in fiber development.
Of the 70,478 predicted protein-coding genes, those with an FPKM >1 in the fiber developmental stages from 5 to 25 DPA were considered to be expressed. Differentially expressed genes (DEGs) between two conditions/groups were analyzed using the DEGSeq R package (1.12.0) (http://bioinfo.au.tsinghua.edu.cn/software/degseq)80. The P values were adjusted using the BH method81. Corrected P values of 0.005, and log2 (fold-change) values of 1, were set as the threshold for significant differential expression.
Assignment of MapMan Bins and GO Terms.
Functional enrichment was analyzed using Mapman82. MapMan bins of the TM-1 genome were assigned by the Mercator pipeline for automated sequence annotation (http://mapman.gabipd.org/web/guest/app/mercator). GO analyses were conducted using Blast2GO83. Within Blast2GO, CDS sequences were used as queries against the nonredundant database in NCBI using BLASTX (E-value 10−6). GO enrichment was derived with Fisher's exact test with the cutoff set at P < 0.05.
Accession codes.
SRA: PRJNA248163. The assembly genome will be available on the laboratory website (http://mascotton.njau.edu.cn) and in the CottonGen database (http://www.cottongen.org).
Accession codes
Primary accessions
Sequence Read Archive
Referenced accessions
Sequence Read Archive
References
Chen, Z.J. et al. Toward sequencing cotton (Gossypium) genomes. Plant Physiol. 145, 1303–1310 (2007).
Li, F. et al. Genome sequence of the cultivated cotton Gossypium arboreum. Nat. Genet. 46, 567–572 (2014).
Wang, K. et al. The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 44, 1098–1103 (2012).
Paterson, A.H. et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 492, 423–427 (2012).
Wendel, J.F. New World tetraploid cottons contain Old World cytoplasm. Proc. Natl. Acad. Sci. USA 86, 4132–4136 (1989).
International Wheat Genome Sequencing Consortium. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345, 1251788 (2014).
Wang, X. et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–1039 (2011).
Liu, S. et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 5, 3930 (2014).
Chalhoub, B. et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345, 950 (2014).
Sierro, N. et al. The tobacco genome sequence and its comparison with those of tomato and potato. Nat. Commun. 5, 3833 (2014).
Kohel, R., Richmond, T. & Lewis, C. Texas marker-1. Description of a genetic standard for Gossypium hirsutum L. Crop Sci. 10, 670–671 (1970).
Li, R. et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272 (2010).
Guo, W. et al. A preliminary analysis of genome structure and composition in Gossypium hirsutum. BMC Genomics 9, 314 (2008).
Wang, K. et al. Localization of high level of sequence conservation and divergence regions in cotton. Theor. Appl. Genet. 124, 1173–1182 (2012).
Wang, K. et al. Structure and size variations between 12A and 12D homoeologous chromosomes based on high-resolution cytogenetic map in allotetraploid cotton. Chromosoma 119, 255–266 (2010).
Jin, J., Zhang, H., Kong, L., Gao, G. & Luo, J. PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 42, D1182–D1187 (2014).
Brenchley, R. et al. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 491, 705–710 (2012).
Otto, S.P. The evolutionary consequences of polyploidy. Cell 131, 452–462 (2007).
Soltis, P.S. & Soltis, D.E. The role of hybridization in plant speciation. Annu. Rev. Plant Biol. 60, 561–588 (2009).
Woodhouse, M.R. et al. Following tetraploidy in maize, a short deletion mechanism removed genes preferentially from one of the two homeologs. PLoS Biol. 8, e1000409 (2010).
Feldman, M. et al. Rapid elimination of low-copy DNA sequences in polyploid wheat: a possible mechanism for differentiation of homoeologous chromosomes. Genetics 147, 1381–1387 (1997).
Gaeta, R.T. et al. Genomic changes in resynthesized Brassica napus and their effect on gene expression and phenotype. Plant Cell 19, 3403–3417 (2007).
Buggs, R.J. et al. Rapid, repeated, and clustered loss of duplicate genes in allopolyploid plant populations of independent origin. Curr. Biol. 22, 248–252 (2012).
Liu, B. et al. Polyploid formation in cotton is not accompanied by rapid genomic changes. Genome 44, 321–330 (2001).
Wang, J. et al. Genomewide nonadditive gene regulation in Arabidopsis allotetraploids. Genetics 172, 507–517 (2006).
Endrizzi, J., Turcotte, E. & Kohel, R. Genetics, cytology and evolution of Gossypium. Adv. Genet. 23, 271–375 (1985).
Motamayor, J.C. et al. The genome sequence of the most widely cultivated cacao type and its use to identify candidate genes regulating pod color. Genome Biol. 14, R53 (2013).
Li, F. et al. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. doi:10.1038/nbt.3208 (20 April 2015).
Gan, X. et al. Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature 477, 419–423 (2011).
Grover, C.E. et al. Homoeolog expression bias and expression level dominance in allopolyploids. New Phytol. 196, 966–971 (2012).
Leach, L.J. et al. Patterns of homoeologous gene expression shown by RNA sequencing in hexaploid bread wheat. BMC Genomics 15, 276 (2014).
Cheng, F. et al. Biased gene fractionation and dominant gene expression among the subgenomes of Brassica rapa. PLoS ONE 7, e36442 (2012).
Kagale, S. et al. The emerging biofuel crop Camelina sativa retains a highly undifferentiated hexaploid genome structure. Nat. Commun. 5, 3706 (2014).
Yoo, M.-J. & Wendel, J.F. Comparative evolutionary and development dynamics of the cotton (Gossypium hirsutum) fibre transcriptome. PLoS Genet. 10, e1004073 (2014).
Adams, K.L., Cronn, R., Percifield, R. & Wendel, J.F. Genes duplicated by polyploidy show unequal contributions to the transcriptome and organ-specific reciprocal silencing. Proc. Natl. Acad. Sci. USA 100, 4649–4654 (2003).
Guan, X., Song, Q. & Chen, Z.J. Polyploidy and small RNA regulation of cotton fibre development. Trends Plant Sci. 19, 516–528 (2014).
Walford, S.A., Wu, Y., Llewellyn, D.J. & Dennis, E.S. GhMYB25-like: a key factor in early cotton fibre development. Plant J. 65, 785–797 (2011).
Wang, S. et al. Control of plant trichome development by a cotton fibre MYB gene. Plant Cell 16, 2323–2334 (2004).
Yang, Z. & Nielsen, R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol. Biol. Evol. 19, 908–917 (2002).
Qin, Y.M. & Zhu, Y.X. How cotton fibres elongate: a tale of linear cell-growth mode. Curr. Opin. Plant Biol. 14, 106–111 (2011).
Haigler, C.H., Betancur, L., Stiff, M.R. & Tuttle, J.R. Cotton fibre: a powerful single-cell model for cell wall and cellulose research. Front. Plant Sci. 3, 104 (2012).
Gou, J.Y., Wang, L.J., Chen, S.P., Hu, W.L. & Chen, X.Y. Gene expression and metabolite profiles of cotton fibre during cell elongation and secondary cell wall synthesis. Cell Res. 17, 422–434 (2007).
Li, X.R., Wang, L. & Ruan, Y.L. Developmental and molecular physiological evidence for the role of phosphoenolpyruvate carboxylase in rapid cotton fibre elongation. J. Exp. Bot. 61, 287–295 (2010).
Broché, M. et al. Transcriptomics and functional genomics of ROS-induced cell death regulation by RADICAL-INDUCED CELL DEATH1. PLoS Genet. 10, e1004112 (2014).
Rodrigues, S.M. et al. Arabidopsis and tobacco plants ectopically expressing the soybean antiquitin-like ALDH7 gene display enhanced tolerance to drought, salinity, and oxidative stress. J. Exp. Bot. 57, 1909–1918 (2006).
Puranik, S. et al. NAC proteins: regulation and role in stress tolerance. Trends Plant Sci. 17, 369–381 (2012).
Turcotte, E. & Feaster, C.V. Semigametic production of haploids in Pima cotton. Crop Sci. 9, 653–655 (1969).
Kim, E.B. et al. Genome sequencing reveals insights into physiology and longevity of the naked mole rat. Nature 479, 223–227 (2011).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Voorrips, R.E. MapChart: software for the graphical presentation of linkage maps and QTLs. J. Hered. 93, 77–78 (2002).
Ooijen, J.W. & Voorrips, R.E. JoinMap® version 3.0: software for the calculation of genetic linkage maps. Plant Res. Inter. (2001).
Kent, W.J., Baertsh, R., Hinrichs, A., Miller, W. & Haussler, D. Evolution's cauldron: duplication, deletion, and rearrangement in the mouse and human genomes. Proc. Natl. Acad. Sci. USA 100, 11484–11489 (2003).
Goodstein, D.M. et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186 (2012).
Altschul, S.F., Gish, W., Miller, W., Myers, E.W. & Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Yu, X.J. et al. Detecting lineage-specific adaptive evolution of brain-expressed genes in human using rhesus macaque as outgroup. Genomics 88, 745–751 (2006).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res. 14, 988–995 (2004).
Stanke, M., Schoffmann, O., Morgenstern, B. & Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinformatics 7, 62 (2006).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94 (1997).
Majoros, W.H., Pertea, M. & Salzberg, S.L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Guigó, R. Assembling genes from predicted exons in linear time with dynamic programming. J. Comput. Biol. 5, 681–702 (1998).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Trapnell, C., Pachter, L. & Salzberg, S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Haas, B.J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Punta, M. et al. The Pfam protein families database. Nucleic Acids Res. 40, D290–D301 (2012).
Hunter, S. et al. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res. 40, D306–D312 (2012).
Altschul, S.F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence data bank and its supplement TrEMBL in 1999. Nucleic Acids Res. 27, 49–54 (1999).
Finn, R.D., Clements, J. & Eddy, S.R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 (2011).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 (2000).
Kanehisa, M. et al. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, D199–D205 (2014).
Lowe, T.M. & Eddy, S.R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, D121–D124 (2005).
Nawrocki, E.P., Kolbe, D.L. & Eddy, S.R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337 (2009).
Nussbaumer, T. et al. MIPS PlantsDB: a database framework for comparative plant genome research. Nucleic Acids Res. 41, D1144–D1151 (2013).
Senchina, D.S. et al. Rate variation among nuclear genes and the age of polyploidy in Gossypium. Mol. Biol. Evol. 20, 633–643 (2003).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Zhang, J., Nielsen, R. & Yang, Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 22, 2472–2479 (2005).
Wang, L., Feng, Z., Wang, X., Wang, X. & Zhang, X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138 (2010).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B. Met. 57, 289–300 (1995).
Thimm, O. et al. MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 37, 914–939 (2004).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Acknowledgements
This work was financially supported in part by grants from the Major State Basic Research Development Program of China (973 Program) (2011CB109300), National Natural Science Foundation of China (No. 3133058; No. 31290213), NSF (ISO1025947), The Fundamental Research Funds by the Central Universities (KYZ201201), Cotton Incorporated (07-161, 14-371), the Priority Academic Program Development of Jiangsu Higher Education Institutions and the 111 program.
Author information
Authors and Affiliations
Contributions
T.Z.Z., Z.J.C. and W.Z.G. conceptualized the research program. T.Z.Z., Z.J.C., R.Q.L., Y.H., X.Y.C. and W.Z.G. designed experiments and coordinated the project. Y.H., L.F., C.X.L., S.W., L.J.C., G.F.M. and D.W.W. prepared DNA sequencing and RNA-seq. X.Y.G., A.M.H.-K., C.A.S., B.E.S., D.M.S., Q.X.S., R.C.K., E.D., D.J.L., D.G.P., P.T. and D.C.J. built BAC libraries and carried out BAC end sequencing. R.Q.L., W.K.J., J.D.C., J.B.Z. and Q.Y.Z. performed assembly and genome annotation. R.Q.L., W.K.J., L.F., J.D.C., J.B.Z., Q. Wan, C.X.L., B.L.L., H.T.W., L.Z., Q.Y.Z., L.N.T., J.L. and Y.L. performed bioinformatics analyses. W.Z.G., T.Z.Z., Y.H., L.F., X.Y.G., J.D.C., M.Q.P., Y.K.W., W.X.Y., W.P.Z., Q. Wang, X.Y.X., H.Z., S.Q.C., Y.T., D.X., X.H.L., J.D., L.N.T., Y.C.L., J.L., Y.L., Z.S.C., C.P.C., X.F.Z. and B.L.Z. were involved in data analysis. T.Z.Z., Z.J.C., R.Q.L., W.Z.G., Y.H., Z.J. and Y.Y.H. supervised the data generation and analysis. T.Z.Z., Z.J.C., Y.H., L.F., X.Y.G., X.Y.C., W.Z.G. and R.Q.L. wrote the manuscript. All authors discussed results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Estimation of TM-1 genome size.
31-mers depth distribution of 180 Gb raw sequence data from libraries with insert size less than 800 bp. Two peaks are observed (49× and 104×): the first peak represents the unique part of the genome and the second peak represents the repetitive part of the genome.
Supplementary Figure 2 Validation of the TM-1 genome assembly using reads mapping.
The alignment information of reads mapping to the TM-1 genome assembly using Burrows-Wheel Alignment (BWA) Tools. X-axis denotes the sequencing depth. Y-axis denotes the fraction of bases.
Supplementary Figure 3 Assessment of the TM-1 genome assembly by four fully sequenced TM-1 BACs.
The red line indicates each BAC sequence and the blue line represents the scaffold. The blocks in white indicate the unfilled gaps on the scaffolds. The orange blocks show aligned region between BAC sequences and scaffolds.
Supplementary Figure 4 Validation of the assembly of A12 and D12 homoeologous chromosomes.
(a) Validation of the assembly using integrated cytogenetic maps of A12 and D12 homoeologous chromosomes. Yellow lines, synthenic gene pairs; red circle, physical position of BAC on chromosome A12 and D12; black bars, relative position of BAC on cytogenetic map of A12 and D1214. Red text indicates centromere. (b) Comparison of the assembly of A12 and G. arboreum chromosome 062.
Supplementary Figure 6 Whole-genome alignments of the G. hirsutum genome and two diploid genomes.
(a) Whole-genome alignments of the G. hirsutum genome using G. raimondii (JGI)4. (b) Whole-genome alignments of the G. hirsutum genome using G. raimondii (BGI-Shenzhen, BGI-SZ)3. (c) Whole-genome alignments of the G. hirsutum genome using G. arboreum (BGI-SZ)2. JGI, Joint Genome Institute. Each of the 13 chromosomes of A and D genomes is designated by color. White blocks represent region which cannot be aligned and only syntenic blocks of ≥ 200 kb in length are shown.
Supplementary Figure 7 Syntenic relationships among T. cacao, G. raimondii4, and G. hirsutum.
(a) Syntenic relationships between T. cacao and G. raimondii4. Notably, the cotton ancestor genome has undergone five to six rounds of whole-genome duplication since its divergence from T. cacao, as shown in the alignment between cacao–cotton orthologous regions. (b) Syntenic relationships between G. raimondii4 and G. hirsutum. Macrosynteny connecting blocks of >30 genes, and numbered rectangles represent the chromosomes.
Supplementary Figure 8 Synteny of the A and D subgenome framework of TM-1 to the D genome4.
(a) Horizontal and vertical axis indicated 13 chromosomes in the A sugenome and D genome4, respectively. (b) Horizontal and vertical axis indicated 13 chromosomes in the D subgenome and D genome4, respectively. A BLASTP search (with an E-value cutoff of 1 × 10−5) was performed to identify paralogous genes.
Supplementary Figure 9 Comparison of codon substitution rate distributions between two subgenomes of TM-1 and their progenitors.
(a) Phylogeny of the cotton genomes using a total of 6,911 orthologous gene families. (b) The comparison of Ka distribution between subgenomes and their corresponding progenitor genomes. (c) The comparison of Ks distribution between subgenomes and their corresponding progenitor genomes. (d) The comparison of Ka/Ks distribution among subgenomes and their corresponding progenitor genomes. All estimations with Ks < 0.01 were removed from the analysis. The central line for each box plot indicated median. The top and bottom edges of the box indicated the 25th and 75th percentiles and the whiskers extend 1.5 times of the interquartile range beyond the edges of the box. The asterisks represented significant difference (Wilcoxon rank sum test, P < 0.05).
Supplementary Figure 10 Distribution of TE divergence level in the A genome2, D genome4 and two subgenomes of TM-1.
Relative proportions of DNA transposons (red), unclassified LTR elements (green), Copia elements (light blue), Gypsy elements (dark blue) and unclassified transposons (grey) are displayed as stacked bar chart.
Supplementary Figure 11 Expression level of disrupted genes versus other genes in TM-1.
Comparison of disrupted genes versus other genes in the A subgenome of TM-1 (left) and D subgenome of TM-1 (right). Average expression levels of the disrupted genes were significantly lower than those of all other genes in two subgenomes (Wilcoxon rank sum test, P < 0.01). The central line for each box plot indicated median. The top and bottom edges of the box indicated the 25th and 75th percentiles and the whiskers extend 1.5 times of the interquartile range beyond the edges of the box.
Supplementary Figure 12 Summary of the number of the expressed genes and average expression levels in all 35 tissues.
Statistics of expression levels of the expressed genes in the different tissues and developmental stages. Blue, 1≤FPKM<10; Orange, 10≤FPKM<50; Green, 50≤FPKM; Dark blue, average expression level.
Supplementary Figure 13 Global comparison of 25,358 homoeologous genes between the two subgenomes of TM-1.
To analyze the global expression pattern of the two subgenomes, the average FPKM values of 25,358 homoeologs and the expressed tissue numbers among 35 tissues. No significant difference on gene expression was found between the two subgenomes of TM-1 (Wilcoxon rank sum test, P > 0.5). Error bars represent s. d.
Supplementary Figure 14 Summary of biased-expressed homoeologous genes in all 35 tissues.
Bar indicated biased-expressed homoeologous gene pairs in each of 35 tissues. The number of expressed genes biased toward the D subgenome was slightly more than those biased toward the A subgenome consistently through most of the tissues. Significant difference is represented by asterisks using Fisher’s exact test (P < 0.05).
Supplementary Figure 15 Summary of the biased homoeologous TFs in all 35 tissues.
(a) Bar indicated the expressed biased homoeologous TF pairs in each of 35 tissues. (b) Number of total bias genes and TFs (MYB and MYB-related type) at 10 and 20 DPA. Significant difference is represented in tissues by asterisks using Fisher’s exact test (P < 0.05).
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–17 and Supplementary Tables 1–25 (PDF 1541 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution- NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/.
About this article

Cite this article
Zhang, T., Hu, Y., Jiang, W. et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat Biotechnol 33, 531–537 (2015). https://doi.org/10.1038/nbt.3207
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nbt.3207
This article is cited by
-
Genome-wide identification of the key Kinesin genes during fiber and boll development in upland cotton (Gossypium hirsutum L)
Molecular Genetics and Genomics (2024)
-
Genomic study of PIFs in cotton and functional analysis of GhPIF4 in promoting cotton flowering
Plant Growth Regulation (2024)
-
Mapping of Genomic Regions for Biochemical and Physiological Parameters Contributing Towards Drought Tolerance in Horsegram (Macrotyloma uniflorum (Lam.) Verdc.)
Applied Biochemistry and Biotechnology (2024)
-
Comprehensive identification and functional characterization of GhpPLA gene family in reproductive organ development
BMC Plant Biology (2023)
-
Lint percentage and boll weight QTLs in three excellent upland cotton (Gossypium hirsutum): ZR014121, CCRI60, and EZ60
BMC Plant Biology (2023)
