
Abstract
Lassa virus (LASV) is a human pathogen, causing substantial morbidity and mortality1,2. Similar to other Arenaviridae, it presents a class-I spike complex on its surface that facilitates cell entry. The virus’s cellular receptor is matriglycan, a linear carbohydrate that is present on α-dystroglycan3,4, but the molecular mechanism that LASV uses to recognize this glycan is unknown. In addition, LASV and other arenaviruses have a unique signal peptide that forms an integral and functionally important part of the mature spike5,6,7,8; yet the structure, function and topology of the signal peptide in the membrane remain uncertain9,10,11. Here we solve the structure of a complete native LASV spike complex, finding that the signal peptide crosses the membrane once and that its amino terminus is located in the extracellular region. Together with a double-sided domain-switching mechanism, the signal peptide helps to stabilize the spike complex in its native conformation. This structure reveals that the LASV spike complex is preloaded with matriglycan, suggesting the mechanism of binding and rationalizing receptor recognition by α-dystroglycan-tropic arenaviruses. This discovery further informs us about the mechanism of viral egress and may facilitate the rational design of novel therapeutics that exploit this binding site.
Similar content being viewed by others


Main
Lassa haemorrhagic fever (LF) is a devastating disease caused by LASV infection. LASV is endemic to West Africa but has spread to new territories in recent years2. Although zoonotic transmission from rodents is the main route of infection, a substantial number of cases have resulted from human-to-human transmission, increasing the risk of large outbreaks. So far, the only therapy for Lassa haemorrhagic fever approved by the US Food and Drug Administration is ribavirin, but this treatment has adverse effects and its efficacy is uncertain12. Thus, there is an urgent need to fill the gaps in our knowledge regarding this lethal pathogen to guide the development of improved therapies.
The spike complex of all arenaviruses undergoes substantial cellular processing before reaching its mature form. The protomers that constitute the homotrimeric complex are first expressed as glycoprotein precursor proteins (GPCs) and are then cleaved twice by a signal peptidase (SPase) and a subtilisin kexin isozyme-1/site-1 (SKI-1) protease. This processing yields three functional subunits: the stable signal peptide (SSP), glycoprotein 1 (GP1) and glycoprotein 2 (GP2) (Fig. 1a). GP1 is the receptor-binding domain that mediates attachment to the cell receptor. GP2 is the transmembrane domain that is responsible for host and viral membrane fusion. Unique to arenaviruses, the SSP translocates with the spike, forms part of the mature complex, can be added in trans, and is important for the maturation and function of the spike5,6,7,8. Despite its critical role in generating a functional spike, it is unknown how the SSP interacts with the other GPC subunits. Although the general architecture of the spike is common to all arenaviruses, different viruses utilize distinct cellular receptors: arenaviruses that infect mammals (mammarenaviruses) can be classified into two serogroups: the Old World (OW) and the New World (NW) groups. Clades B and D of NW arenaviruses use transferrin receptor protein 1 (TfR1) as a cellular receptor13,14; Lujo virus, which differs from the OW and NW viruses, uses neuropilin-215 (NRP2); and OW viruses such as LASV and clade-C NW arenaviruses use α-dystroglycan3,4 (α-DG). Recognition of α-DG depends on matriglycan16, which is a unique linear carbohydrate modification synthesized by LARGE117. Matriglycan comprises repeating units of xylose (Xyl) and glucuronic acid18 (GlcA) that are linked together in the form of [-3Xyl–α1,3-GlcA-β1-]n. The linear chains of matriglycan consist of many such repeats, enabling them to project to substantial distances from the cell surface.

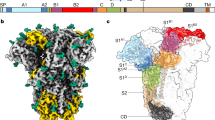
a, A schematic view of the spike complex subunit organization. b, Overview of the structure. Top left, LASV spike electron density map. The trimeric spike is shown with a different colour (blue, grey or pink) for each protomer and with different tones for the SSP, GP1 and GP2 subunits. N-linked glycans are shown as spheres. The rough boundaries of the membrane bilayer and the visible termini are indicated. c, Organization of the transmembrane helices. The six transmembrane helices are shown along the three-fold symmetry axis of the spike (marked with a triangle) from an intracellular viewpoint. d, Energy profiles (in Rosetta energy units (REU)) for the different registers of SSP. This graph shows a focused view of registers that place the first hydrophobic region of SSP, or part of it, in the membrane. Blue and red lines indicate threading the SSP with its C terminus out and N terminus out, respectively. The lowest-energy global solution is indicated with a large red dot. e, Side view of the transmembrane helices indicates their membrane-crossing angle. f, Close-up view of the SSP. One SSP is shown as a green ribbon, and the rest of the spike is shown as a surface coloured by the electrostatic potential: blue, 5 kT e−1; white, 0; red, −5 kT e−1. g, Multiple sequence alignment of the SSPs from the indicated viruses. The first (h1) and second (h2) helices of SSP are indicated as well as the extracellular region. The transmembrane region is shown in red and the unmodelled cytoplasmic tail is in grey. Previously identified hydrophobic regions are noted. Fully conserved residues are highlighted with a yellow background. DANV, Dandenong virus; GTOV, Guanarito virus; JUNV, Junín virus; LATV, Latino virus; MORV, Morogoro virus; OLVV, Oliveros virus; WWAV, Whitewater Arroyo virus.
Although it is known how TfR1 and NRP2 are recognized by arenaviruses19,20, the structural basis for α-DG recognition remains unknown. An important step was made by Hastie et al., who determined the structure of a mutationally stabilized ectodomain of the LASV spike complex21. This enabled the mapping of residues that were previously found to influence binding to α-DG by the OW lymphocytic choriomeningitis virus22,23,24 (LCMV) onto the LASV spike. One of these residues, Tyr150, was found at the interface between two adjacent GP1 subunits at the apex of the spike, but the actual mechanism for binding α-DG remained unknown. Other structural studies on components of the LASV spike complex and related proteins25,26 have provided further clues regarding the basis of receptor recognition, yet key questions remain. Here we address these questions using a more holistic approach, investigating the structure of the complete, membrane-embedded, spike complex carrying only minimal modifications. We present the cryo-electron microscopy (cryo-EM) structure of the LASV spike complex, which provides fundamental information regarding the SSP, receptor recognition and the stability of this trimeric complex.
Topology and structure of the SSP
To determine the structure of the LASV spike complex, we produced the full-length GPC of LASV fused to a C-terminal Flag tag in HEK 293F cells. Single-particle cryo-EM analysis using detergent-solubilized, purified spike provided a three-fold symmetric density (Extended Data Figs. 1, 2). Gold-standard Fourier shell correlation (GSFSC) indicated a resolution of 2.5 Å (Extended Data Fig. 1), but the map quality suggested a lower resolution, and the map model Fourier shell correlation (FSC) extended to 3.3 Å (Extended Data Fig. 2). We thus low-pass filtered the map to 3.3 Å to avoid high-frequency noise. The map reveals that the spike has six transmembrane helices organized in a bundle (Fig. 1b, Extended Data Fig. 1). We used the available structure of the LASV ectodomain21 (Protein Data Bank (PDB): 5VK2) as a starting model, and manually completed and refined the model (Fig. 1b, Extended Data Fig. 2, Extended Data Table 1). The membrane domain is organized such that the central three helices belong to GP2 subunits (Fig. 1a, c), and the outer three helices belong to the SSPs (Fig. 1c). Modelling the central GP2-helices was straightforward, as their register is dictated by the extracellular portion of the spike. Density for side chains of the SSP transmembrane helices point toward the extracellular side (Extended Data Fig. 3a), a directionality that is apparent even in the low-pass-filtered map (Extended Data Fig. 3b), clearly establishing that the N terminus is extracellular.
We were further able to trace the main chain of about 12 residues of SSP outside the membrane. However, the quality of the density in this region was not sufficient to unambiguously determine the register. To address this, we used an unbiased computational modelling approach using Rosetta all-atom modelling. First, the transmembrane region was embedded in a virtual membrane. Then, we threaded all possible registers of SSP (37 possibilities for each direction) on the 6-helix bundle, while maintaining the register of the central GP2-helices. In each modelling trajectory, the sidechain conformations were packed and the sidechains and backbone were minimized using an energy function dominated by van der Waals interactions, hydrogen bonds, electrostatic forces and membrane-depth-dependent solvation27 (Extended Data Fig. 3c). Placing the first hydrophobic region of the SSP in the membrane provided the lowest-energy model, with a significant energy gap compared to all other possibilities (Fig. 1d, Extended Data Fig. 3c). Models with threaded sequences in reverse (that is, with the C terminus outside) exhibited much higher energies overall (Fig. 1d, Extended Data Fig. 3c). This analysis favoured a single solution (Extended Data Fig. 3d) which we then adopted for completion of our model.
The SSP starts with a short helical segment (h1) that lies parallel to and is probably partially embedded in the membrane (Fig. 1b) before its transmembrane helix enters the membrane at an angle of around 70° (Fig. 1b, e). Hydrophobic residues in h1—such as Ile4—seem to face and probably penetrate into the membrane (Fig. 1f). The highly conserved Pro12 serves as a helix breaker and seems to terminate h1 (Fig. 1b, f). Glu16 and the conserved Glu17 are predicted to face a positively charged patch that is formed by residues in GP2, and potentially interact with it (Fig. 1f, g). Lys33, which is highly conserved and was previously linked to pH-induced membrane fusion8, is located at the very end of the transmembrane helix of SSP (Fig. 1f) and is either exposed to, or snorkels with its side-chain toward the cytoplasm.
Previous experimental studies corroborate our model of SSP. Specifically, the conserved Pro12 is important for allowing the break between h1 and h2, and other residues may disfavour the observed bound conformation of SSP. Indeed, mutating Pro12 in LCMV spike (Fig. 1g) to either alanine or glycine does not affect the surface expression of the protein, but completely abrogates infectivity of pseudotyped viruses28, suggesting a premature dissociation of the SSP from the spike. Furthermore, in our structure, Glu16 and Glu17 of SSP point toward a positively charged patch on GP2 (Fig. 1f). Mutating the equivalent negatively charged residues of LCMV (Asp16 and Glu17) to alanine does not affect surface expression but, as in the case of Pro12, abrogates the infectivity of pseudotyped viruses28. Reverting the charge of these two residues by mutating them to lysine (that is, D16K/E17K) results in a more deleterious effect that reduces surface expression and maturation of the spike on top of abrogating infectivity of pseudotyped viruses28. This indicates that SSP has reduced association with the spike even inside the endoplasmic reticulum–Golgi network, since this interaction is needed for the proper maturation of the spike6. Our structure explains not only the deleterious effects, but also the lack of effect of mutating some residues. Specifically, mutating either Gln3 or Thr6 of LCMV (corresponding to Gln3 and Val6 in LASV) to alanine does not have any functional consequence for the spike28. Both of these SSP residues are predicted by our structure to be solvent-exposed (Fig. 1f), rationalizing this observation.
Different models for the topology of SSP have been proposed9,10,11,29, including a bitopic model in which the two hydrophobic regions cross the membrane9. Our structural data, however, indicate the presence of only six transmembrane helices (Fig. 1c, Extended Data Fig. 2), and not nine as predicted by the bitopic model. Further, our structure indicates that the N terminus of SSP is located on the outer side of the membrane (Fig. 1e, Extended Data Fig. 3a), which also differs from the bitopic model9. Although the observed configuration of the SSP does not support the previously proposed models, it is consistent with some published biochemical data. Specifically, Cys57 was shown to participate in zinc coordination together with the cytoplasmic tail of GP230, an observation that is consistent with the observed topology of SSP in our structure. Furthermore, it has been demonstrated that the first hydrophobic region of the SSP interacts with GP2 inside the membrane31, which is also consistent with our structure. Last, our structure indicates that the positively charged residues of SSP are indeed on the cytoplasmic side of the membrane (Fig. 1g), an organization that is favoured32 (that is, the ‘positive inside rule’).
Double-sided stabilization of the spike
The modified ectodomain of LASV was found to be labile and unstable in the absence of ‘stapling’ antibodies21. This observation was surprising considering that LASV is transmitted through aerosolized animal excreta and should therefore withstand harsh environmental conditions. By contrast, the detergent-solubilized native spike is stable without the addition of any binding partners. Several structural factors contribute to this apparent stability; at the apex of the trimer, the C termini of the GP1 subunits are mutually swapped and interlocked with the neighbouring protomers (Fig. 2a). This GP1 region contains the RRLL recognition motif of SKI-I33 (Fig. 1a). It engages a neighbouring GP1 subunit via hydrophobic interactions through Ile254, Leu258 and Leu259, as well as polar interactions between Arg257 and 3 main-chain carbonyl groups (Fig. 2a). Tyr150 is an important residue for this interaction, as it is engulfed by the aliphatic part of Arg257 and by Leu258 (Fig. 2a). Leu258 reaches to and interacts with the interface between the two other GP1 subunits (Fig. 2a).

a, A domain swap at the apex of the trimer. A top view of the trimer using a surface representation for the three individual GP1 subunits, shown in pink, blue and grey. Inset, magnified view of the domain-swapped element. Key interacting residues are labelled and shown as sticks. Yellow dashed lines represent polar interactions between the Arg guanidino group and main-chain carbonyl oxygens. b, Domain swapping at the membrane-proximal region of the trimer. Main image, the three GP2 subunits are shown using ribbon and surface representations. The SSPs were omitted for clarity. The transmembrane (TM) region is indicated. Bottom left, the organization of secondary structures in GP2. Right, magnified view of the interaction between the GP2 subunits. Key interacting residues are labelled and shown as sticks. Yellow dashed lines represent polar interactions. c, A similar view as in b, right, with the addition of one SSP.
On the opposite side of the trimer, at the membrane-proximal region, we found an additional domain swap that involves the GP2 subunits. The transmembrane helix of each of the GP2 subunits does not penetrate the membrane in proximity to its cognate subunit. Instead, a long α-helix (h5) is projected toward its neighbouring GP2, interacts with the adjacent h5 helix, and penetrates into the membrane such that the transmembrane helices are twisted around each other (Fig. 2b). The h5–h5 interaction includes hydrophobic contacts between Leu415 and Pro427 (which also serves as a helix breaker) and between Tyr419 and the aliphatic part of Gln423 (Fig. 2b). In addition, Gln416 makes a hydrogen bond with the main-chain carbonyl of Met420, and Arg422 makes a salt bridge with Asp432 (Fig. 2b). The mutual h5-swapping of the GP2s is locked in place by the SSPs that prevent the h6 transmembrane helices of GP2 from unwinding (Fig. 2c). The transmembrane helices of the SSP are in grooves between each two central GP2 helices, forming multiple hydrophobic interactions (Fig. 1b, c, e). The extracellular h1 of SSP (Fig. 1b) makes hydrophobic interactions with its cognate GP2 h5 and a neighbouring GP2 h6 through Phe7 and Val11 (Fig. 2c). In addition, Glu16 of the SSP forms a polar interaction with Arg422 of the neighboring GP2 h5 (Fig. 2c). Of note, the mutual swapping at the apex and at the membrane-proximal region of the spike have opposite directionalities (Extended Data Fig. 4), in a configuration that could be envisioned as a sweet wrapper with two twisted ends.
Recognition of matriglycan by LASV
A key open question is how matriglycan-dependent recognition of α-DG is achieved. While solving the structure, we noticed elongated density blobs in deep pockets formed by the C terminus of GP1 and Tyr150 (Fig. 3a). These densities do not resemble in size and shape any of the compounds used for producing the spike. The proximity to Tyr150, which influences α-DG binding24, suggests that these densities may belong to matriglycan. Indeed, segments of matriglycan [-Xyl–α1,3-GlcA–β1,3-Xyl-] moieties readily fit into these density blobs (Fig. 3a). We generated a locally focused C3-symmetric map (Extended Data Fig. 5a) to provide a more detailed view of these densities (Fig. 3a). The size and shape of the densities agree well with the model of matriglycan. To confirm the presence of matriglycan, we used the IIH6 anti-matriglycan antibody34. Detergent-solubilized LASV spike that is captured on anti-Flag beads is able to pull down IIH6, whereas the spike of the TfR1-tropic arenavirus Machupo virus13 (MACV) cannot (Fig. 3a, Extended Data Fig. 6a). The presence of matriglycan is further evident in MLV-based pseudotyped viruses that bear the spike complexes of LASV but not of the neuropilin-2-tropic15,20 LUJV (Extended Data Fig. 6b).

a, Left, top view showing the apex of the trimer using surface representation for the different GP1 subunits (pink, blue and grey). N-linked glycans are shown as spheres. Blobs of density next to Tyr150 are shown as a blue mesh at σ = 4, carved at 2 Å around the Xyl–α1,3-GlcA–β1,3-Xyl moieties, which are shown as sticks. Top right, a closeup view of the density around the Xyl–α1,3-GlcA–β1,3-Xyl moiety (carved at 2 Å, σ = 4) in a focused C3-symmetric map. Bottom right, dot blot analysis of membrane-solubilized Flag-tagged GPCs of LASV and MACV, co-purified in the presence of the anti-matriglycan IIH6 antibody. Top blot shows detection of IIH6; bottom blot shows detection of Flag tag. Representative image from three independent repeats. b, Detailed view of the matriglycan-binding site at two angles, using the same colour scheme as in a. Polar interactions are shown as dashed lines in yellow for spike–matriglycan, in green for inner spike, and in purple for inner matriglycan. Grey dashed line shows hydrophobic interaction. c, A continuous matriglycan chain in a focused C1-symmetric map. Density is shown as a blue mesh (carved at 2 Å, σ = 4.5) around the matriglycan. Colour scheme as a. The Xyl–GlcA–Xyl triad in the symmetric sites is shown in orange. Linker GlcA at the asymmetric sites and the first Xyl (marked with a red asterisk) are in green. N-linked glycans (N-Gly) are shown as spheres. d, Detailed view of the binding of GlcA at the asymmetric sites. Polar interactions are shown as dashed lines. e, Amino-acid frequency plots of the six terminal GP1 residues from α-DG- and TfR1-tropic arenaviruses. AMAV, Amapari virus; CHAPV, Chapare virus; IPPYV, Ippy virus; LUAV, Luna virus; MOPV, Mopeia virus; SABV, Sabiá virus; TCRV, Tacaribe virus.
Matriglycan recognition by the spike is mediated by multiple polar interactions. The central sugar in the pocket is GlcA, and its carboxyl group makes polar interactions with the main-chain amines of Arg257 and Leu258, and a salt bridge with the guanidino group of Arg257 (Fig. 3b). Arg257, which is pre-positioned for binding by the three polar interactions it makes with the main-chain carbonyls, forms additional polar bonds with the ring-forming GlcA-O5, with the glycosidic bond of GlcA–Xyl, and the hydroxyl group on Xyl-C2 (Figs. 2a, 3b). Tyr150 is stabilized by polar interactions with Glu151 and Lys125 from a neighbouring GP1 and forms hydrophobic interactions with GlcA (Fig. 3b), thus rationalizing its previously assigned contribution for binding α-DG in LCMV24 and in LASV35. In addition, Arg256 forms two polar interactions with the hydroxyl group on Xyl-C2. Notably, the observed bound conformation of matriglycan may represent a low-energy conformer, since it is stabilized by several intra-matriglycan polar interactions (Fig. 3b). In addition, the formation of the binding site for matriglycan by a RRLL motif from one GP1 subunit that is packed against another GP1 subunit (Figs. 2a, 3b) explains why monomeric LASV GP1 subunits do not form detectable interactions with α-DG25.
Next to each of the three Xyl–GlcA–Xyl moieties, we observed density that indicated a continuation of the chain but was not sufficiently clear for modelling. We hence further classified our data in C1 and created focused C1-symmetric maps for this region (Extended Data Fig. 5b). The main class clearly showed that the three densities comprise, in fact, parts of a single continuous linear chain of matriglycan (Fig. 3c, Extended Data Fig. 6c). We were able to model linking-GlcA moieties at two of the asymmetric sites (Fig. 3c, Extended Data Fig. 6c, Extended Data Table 1). In addition, we were able to model additional Xyl–GlcA at the entrance point of the matriglycan (Fig. 3c). The terminal Xyl moiety in our model stacks parallel to a N-acetylglucosamine attached to Asn119 (Fig. 3c). All the extra GlcA moieties form favourable contacts with the LASV spike; the carboxyl groups of the GlcA in the asymmetric sites serve as N-terminal caps for α-helices of GP1 by forming hydrogen bonds with the main-chain amines of Leu120 and Ser121 (Fig. 3d). In addition, at least for one linking GlcA, the hydroxyl of Ser121 seems to form a hydrogen bond with the ring oxygen atom and with the carboxyl group of GlcA. In total, we observe 13 sugar monomers bound to the spike of LASV with a total buried surface area of 2,113 Å2 (903 Å2 on the spike and 1,210 Å2 on the matriglycan chain) for complex formation. Thus, extensive network of polar interactions, large buried surface area and additional factors such as degeneracy in binding states, drive the recognition of matriglycan by LASV. Of note, the role of the SKI-I RRLL recognition motif in binding matriglycan, and particularly the contribution of Arg257 (Fig. 3b), explain the conservation of this residue in α-DG-tropic but not in TfR1-tropic arenaviruses (Fig. 3e), a difference that has been noted before33. This structure further explains how mutating His141 and Phe147 to alanine abrogates the interaction of the LASV spike with α-DG35, as these two residues are located directly below the RRLL motif (Extended Data Fig. 7a) and thus contribute to the stabilization of the matriglycan-binding site.
Spike inhibition by free matriglycan
The discovery that the LASV spike complex is generated preloaded with matriglycan raises questions about the source of this molecule and its role in the biology of LASV. Matriglycan is thought to be a unique modification of α-DG16,17. To test whether the LASV-bound matriglycan is attached to α-DG, we spotted a nitrocellulose membrane with detergent-solubilized Flag-tagged LASV spike and probed it with the IIH6 antibody (Fig. 4a). Although matriglycan signal is apparent in a total cell lysate that contains α-DG, no signal was observed in the LASV spike (Fig. 4a), indicating that matriglycan was washed away. This implies that the spike-carried matriglycan is not covalently attached to a protein mass that could have retained it on the membrane, a notion that is further corroborated by the uniform appearance of particles in the electron micrographs (Extended Data Fig. 1) and the lack of unaccounted 2D classes in our data. These results further suggest that cells produce matriglycan that is not covalently linked to α-DG.

a, Dot blot analysis of membrane-immobilized matriglycan. Purified LASV GPC and total cell lysate (TCL) of HEK 293T cells were spotted on a nitrocellulose membrane. The presence of matriglycan (top) was detected using IIH6 antibody. After stripping, the presence of Flag was detected using anti-Flag antibody (bottom). One representative experiment out of three independent repeats. b, Excess of matriglycan reduces infectivity of LASV. Infectivity of MLV-pseudotyped viruses carrying the spike complexes of LASV or of the TfR1-tropic MACV was measured in the presence or in the absence of excess matriglycan. Dots represent technical repeats (n = 5) and P values (two tailed Student’s t-test) are indicated. Whiskers indicate the minimum and maximum values, the central line indicates the mean, and the box shows the interquartile range. One representative experiment out of three independent repeats. c, Schematic model for the effect of soluble matriglycan on the cell-attachment potential of LASV. The red curve represents the decay in the number of spike complexes that are bound to soluble matriglycan following the transition of the viruses into an environment with low free matriglycan concentration (that is, much lower than the KD for complex formation). The cell-attachment potential is indicated by a grey curve, and is cooperatively growing, owing to the high-avidity potential of binding to the target cell. A window of opportunity in which viruses can diffuse before their attachment potential becomes high and causes them to adhere to a target cell is indicated with a blue rectangle.
Soluble free matriglycan that occupies the spike binding site may serve as a competitive inhibitor for binding cells. Indeed, supplementing MLV-based LASV pseudotyped viruses with in vitro-produced matriglycan significantly inhibits infectivity (Fig. 4b). Therefore, once newly formed viruses are exposed to a low free-matriglycan concentration environment (that is, below the dissociation constant (KD) for complex formation), the number of blocked spikes will decay (Fig. 4c) until there would be a sharp increase in the attachment potential of the viruses to cells owing to the high avidity potential of attachment (Fig. 4c). We postulate that under such conditions, newly formed viruses will have a window of opportunity to escape the cell from which they originated before their attachment potential becomes too high. Thus, binding of free matriglycan by the LASV spike probably aids viral egress.
Discussion
The structural role of the unusually long SSP of arenaviruses has so far remained unknown. Owing to the high conservation of SSP in arenaviruses (Fig. 1g), it is highly likely that they all share the same SSP configuration, as we discovered here. Our data indicate that the second hydrophobic region of SSP (Fig. 1g) is not associated with the central transmembrane region of the spike, and is located at the cytoplasm or inside the virion. The organization of the SSP indicates that besides the 38 residues of GP2 that form a cytoplasmic zinc-binding domain30, there are also around 25 residues of SSP inside the cell (Fig. 1f, g). Thus, the spike complexes of arenaviruses have a substantial protein mass located at the intraviral–inner-cellular region. The separation of the C terminus of SSP and the N terminus of GP1 across the membrane indicates a topological rearrangement event that probably occurs after cleavage by SPase. It is noteworthy that topological changes of signal peptides have been observed previously36,37, and this phenomenon might explain the difficulty of experimentally determining the topology of the SSP. The association mode of SSP with the trimeric spike (Fig. 1b) suggests that it helps in maintaining the stability of the native structure by locking in place the transmembrane helices of GP2 (Fig. 2c). We speculate that structural rearrangements of the spike probably occur during priming38 and may involve changes in the way SSP interacts with the spike. This may help rationalizing the role of SSP residues, such as Lys33, in affecting pH-induced triggering of the spike8.
In addition to the multiple polar interactions and the extended buried surface area that drive the binding of matriglycan by the LASV spike complex, other factors may influence the binding strength. The interaction of the LASV spike complex, which exhibits three-fold symmetry and harbours a linear asymmetric form of matriglycan, gives rise to multiple degenerate binding states (Extended Data Fig. 8a). Although these states are fully equivalent and indistinguishable, their existence effectively reduces the entropic cost of binding. Besides the observed conformation in our structure (Fig. 3c), additional states might be accommodated using similar geometry. Two or even three separate matriglycan chains could potentially bind to a single trimer at the same time, and with multiple degenerate states (Extended Data Fig. 8b, c). Simultaneous binding of more than one chain may be important for establishing the initial interaction of the virus with the host cell and should also benefit from avidity if the matriglycan chains are anchored together to the same surface. The spike complex could also possibly bind in the middle of a long matriglycan chain. A proposed geometry for such binding is shown in Extended Data Fig. 8d. This putative interaction may allow the virions to slide along the long matriglycan chains, similarly to sliding of various proteins along other biological polymers39.
The finding that the RRLL–SKI-I recognition motif has important structural roles in stabilizing the spike (Fig. 2a) and in forming the matriglycan-binding site (Fig. 3a), provides an explanation for the evolutionarily conserved function of SKI-I in cleaving the GPC. For other viruses that utilize class-I spike complexes, such as SARS-CoV-2, acquiring a furin cleavage site increases their virulence40,41 by achieving a more efficient cleavage as well as enabling the recognition of the newly formed C′ by NRP1 and NRP2, which become attachment factors41. To enable binding by the NRPs, the newly formed C′ must stay exposed in the mature spike, which is true for SARS-CoV-2, but not for LASV (Extended Data Fig. 9). Artificially introducing a furin site to the spike complex of LASV results in a cleaved trimer21 that lacks the stabilizing swapping mechanism as we observed, and has a malformed receptor binding site (Fig. 2a, Extended Data Fig. 7b).
Viruses are known to ‘borrow’ molecules from their host cells during biogenesis. A known example is the ‘pocket factor’, a cellular-derived fatty acid that binds and stabilizes the coat proteins of rhinoviruses42,43. A recent example includes the spike complex of SARS-CoV-2, which uses linoleic acid from its host cell to stabilize a ‘closed’ conformation44. After maturation, the LASV spike complex latches on to what appears to be a free matriglycan polymer (Fig. 4a,). For this to happen, there are two important prerequisites: (1) the spike complexes need to be able to bind matriglycan, and (2) matriglycan should be available. SKI-I is localized to the Golgi45 and the processing of the LASV GPC by SKI-I occurs at the endoplasmic reticulum–cis-Golgi46. The enzyme that produces matriglycan is LARGE116,18 and is also located at the Golgi47. However, LARGE1 is known to synthesize matriglycan on α-DG and not as a free polymer in solution. Notably, incubating the enzyme β1,4-glucuronyltransferase (B4GAT1)48 with Xyl and UDP-GlcA in vitro produces a GlcA–β1-4-Xyl that is recognized and elongated by LARGE149. Hence, B4GAT1 that is found in the Golgi48 could potentially produce free substrates that LARGE1 would subsequently elongate to form free polymers of matriglycan. We therefore postulate that following SKI-I-depended maturation at the endoplasmic reticulum–cis-Golgi, the LASV spike reaches a Golgi compartment where free matriglycan polymers are available, and that this is where loading occurs. The bound matriglycan then serves as a competitive inhibitor for the binding of matriglycan on cells (Fig. 4b). Inhibiting cell attachment for a certain duration using free matriglycan provides a mechanism that reduces the probability of newly formed α-DG-tropic arenaviruses adhering back to their host cells, and thereby facilitates viral egress. Moreover, since the LASV spike complex undergoes priming in a low-pH endocytic environment during cell entry38, the presence of free matriglycan in the trans-Golgi network—also characterized by low pH50—could help stabilize the spike to reduce premature priming.
Introducing various modifications, such as truncations, point mutations and alterations to viral spike proteins has been an essential approach for obtaining important structural information. Our study demonstrates that such modifications may unintentionally alter essential structural features, such as the formation of the matriglycan-binding site (Extended Data Fig. 7b), as in the case of the LASV spike. Using single-particle cryo-EM may circumvent or at least reduce the need to introduce such modifications. The structure of a stable LASV spike and the identification of the matriglycan-binding site may facilitate the design of improved therapies targeting this high-priority human pathogen.
Methods
LASV GPC production and purification
Expression of the full-length LASV GPC was carried out in HEK 293F cells (Invitrogen) using FreeStyle Medium (Life Technologies). Cells were grown to a density of approximately 1.0 × 106 cells per ml before transfection. HEK 293F cells were transfected using 40 kDa polyethylenamine (PEI-MAX) (Polysciences) at 1 mg ml−1, pH 7 with DNA at a ratio of 1:2.5 (DNA:PEI solution). Codon-optimized LASV GPC (Josiah strain) was chemically synthetized (Genescript) and then subcloned with a C-terminal Flag tag into pcDNA3.1 using BamHI–NotI restriction sites. The LASV GPC-expressing cells were collected at 48 h post-transfection by centrifugation at 600g, 4 °C for 10 min. Membranes were then resuspended in a cold lysis buffer (10 mM Tris, 100 μM MgCl2, 100 μM phenylmethyl sulfonyl fluoride (PMSF), 15% glycerol) and homogenized for 5 min on ice. The lysis mixture was then incubated while rotating for 1 h at 4 °C. A second homogenization was carried out and the lysis mixture was centrifuged at 21,000g for 25 min, 4 °C. The supernatant was discarded and pellets were dissolved in solubilization buffer (20 mM Tris, 150 NaCl, 50 μM ZnCl2, 100 μM PMSF, 15% glycerol, 2% (% w/v) n-dodecyl-β-d-maltoside (DDM; Anatrace), 0.2% (% w/v) cholesteryl hemisuccinate (CHS; Anatrace). The solubilization mixture was then homogenized and incubated for 4 h; after which it was centrifuged at 265,000g for 25 min, 4 °C. The supernatant of this solubilization step was then incubated overnight with 40 μl of EZview red anti-Flag beads (Sigma Aldrich). The insoluble material from the solubilization step was discarded. The anti-Flag beads were then spun down (800g, 1 min) and washed by subsequently decreasing amounts of glycerol to 0.3% while replacing the DDM and CHS with increasing amounts of lauryl maltose neopentyl glycol (LMNG; Anatrace) to 0.03%. The protein was eluted after the last washing step with 120 μl of 0.40 mg/ml of 1× Flag peptide (Genescript) in a buffer containing 0.03% LMNG, 0.3% glycerol, 20 mM Tris-HCl, 150 mM NaCl, 50 μM ZnCl2, 100 μM PMSF, from a 2-hour incubation on ice with periodic mixing. For western blot analysis, anti-Flag primary antibody (Thermo Fischer) was used at 1:1,000 dilution with horseradish peroxidase (HRP)-conjugated anti-rabbit secondary antibody (Jackson) at a 1:10,000 dilution.
Cryo-EM image acquisition, data analysis and 3D reconstruction
Purified LASV GPC sample (3.5 μl) was applied on glow-discharged (10 s, 12 mA; Pelco easiGlow, Ted Pella) graphene oxide Quantifoil copper grids, R1.2/1.3, (Electron Microscopy Sciences) using a Vitrobot system (Thermo Fischer/FEI) (3 s blotting time, 4 °C, 100% humidity). Samples were incubated on the grid for 2 min before blotting was carried out. cryo-EM data was then collected on the Titan Krios microscope (FEI) operated at 300 kV, using a Gatan K3 direct detection camera. The beam size was 900 μm diameter, the exposure rate was 20 e− s−1 pixel−1, and movies were then obtained at 165,000× magnification with a pixel size of 0.519 Å. The nominal defocus range was −0.6 to −1.8 μm. A total of 4,072 movies were automatically collected using EPU.
Data processing was carried out with the cryoSPARC v3 suite51. Patch motion correction and patch CTF estimation were carried using cryoSPARC Live. Blob picking was used to pick 1,208,607 particles. Particles were extracted using a 512-pixel box, and were Fourier-cropped to a 256-pixel box with a pixel size of 1.038 Å. The dataset was cleared using several rounds of 2D classification, followed by 3D classification using C1 symmetry, and ab initio-created models. Some 97,542 particles belonging to a single class with the most visible transmembrane region were selected and further refined using 2D classification to yield a final data set of 91,903 particles that was used for reconstructing the density map while imposing a C3 symmetry. The final map was low-pass filtered to the map-model FSC value (see Extended Data Fig. 2).
To reconstruct focused maps, 234,687 particles belonging to classes 2, 3 and 4 that exhibited an apparent complete ectodomain were pooled together and reconstructed with C3 symmetry followed by local C3-symmetric reconstruction with a mask around the ectodomain (first map). The same pool of particles was further 3D-classified using a C1 symmetry, and a subset of 103,416 particles was used to first reconstruct and then to refine using local reconstruction a C1-symmetric map (second map). The final map was low-pass filtered to the map-model FSC value (see Extended Data Fig. 6c).
Model building, refinement and analysis
An initial model was generated by docking the ectodomain structure obtained by Hastie et al. 21 (5VK2) into the density map using Chimera52. Then, using Coot53 and real-space refinement in Phenix54, we manually completed and refined the model of the spike. We used PyMol55 for structural analysis and generation of graphics. We used Areamol within the CCP4 suite56 to calculate buried surface area.
Structure modelling of the signal peptide
Membrane-spanning segments were identified manually. The structure was embedded in a virtual membrane using the PPM server57. The region encompassing the signal peptide (positions 1–58) was segmented into all possible 22 amino acid windows, generating 37 possible sequences. Each sequence was modelled in Rosetta in an orientation corresponding to a C terminus in the cytoplasm or out. Each sequence was threaded on the backbone coordinates as observed in the experimental structure, followed by side-chain packing and side-chain and backbone minimization. All calculations were done using the membrane protein energy function ref2015_memb27,58, which is dominated by van der Waals interactions, environment-dependent hydrogen bonding, electrostatics, and a depth- and orientation-dependent membrane solvation potential derived from the dsTβL experiment58. Each window was modelled ten times in each orientation, retaining the model with the lowest computed energy for each window. See Supplementary Information for the RosettaScript59 and command line for executing calculations.
UniProtKB accession codes for GPC sequences and viruses abbreviations
The virus name abbreviations and the sequence entry codes for the relevant sequences that were used in this study are: Lassa (LASV, P08669), Dandenong (DANV, B1NX58), Guanarito (GTOV, Q8AYW1), Junín (JUNV, P26313), Latino (LATV, Q8B121), Lymphocytic choriomeningitis (LCMV, Lymphocytic choriomeningitis), Lujo (LUJV, C5ILC1), Morogoro (MORV, C6ZK00), Oliveros (OLVV, Q84168), Mopeia (MOPV, P19240), Ippy (IPPYV, Q27YE4), Luna (LUAV, A0A6B7EQ86), Mobala (MOBV, Q2A069), Sabiá (SABV, Q90037), Chapare (CHAPV, B2C4J0), Amapari (AMAV, Q8B122), Tacaribe (TCRV, P18141), Machupo (MACV, Q6IUF7) and Whitewater Arroyo (WWAV, Q911P0).
Biochemical analysis for the presence of matriglycan
For detecting matriglycan we made use of the anti-matriglycan IIH6 monoclonal antibody. The IIH6 C4 monoclonal antibody developed by K. P. Campbell was obtained from the Developmental Studies Hybridoma Bank (DSHB), created by the NICHD of the NIH and maintained at The University of Iowa. Flag–LASV GPC was solubilized from membranes and immobilized on anti-Flag beads as described above for the purification of the spike. Following a wash at a buffer containing 7.5% glycerol, 50 μl of 30 μg ml−1 IIH6 C4 was added to 250 μl of the anti-Flag beads in a 5% glycerol wash buffer (with 0.04% LMNG and residual DDM). The anti-Flag beads were then incubated for 1 h with IIH6 with intermittent mixing. The remainder of the washing and elution steps were carried out exactly as described for the purification of the spike. Upon elution by the Flag 1× peptide, protein samples were then directly used for immunoblotting on a nitrocellulose membrane. To detect IIH6, we blocked the membrane with 3% skim milk and used goat anti-mouse IgM–HRP (Santa Cruz; 1:2,500 dilution) in TBST with EZ-ECL (Biological Industries) for visualization. For detecting Flag, the membrane was washed with transfer buffer (25 mM Tris-HCl, 200 mM glycine, 20% methanol), blocked with 3% BSA in TBST, incubated with rabbit anti-Flag polyclonal antibody (Thermo Fisher; 1:2,000) for 1 h, and with anti-rabbit HRP-conjugated IgG (Jackson ImmunoResearch Laboratories; 1:10,000), prior to the visualization with EZ-ECL.
Production of pseudotyped virus-like particle
Murine leukaemia virus (MLV)-based, virus-like particles (VLPs) pseudotyped with the GPC spike complexes of LASV, Lujo virus (LUJV) and Machupo virus (MACV) were produced by co-transfecting retroviral transfer vector pLXIN-Luc encoding a luciferase reporter gene together with a pcDNA3.1 encoding either MACV, LUJV or LASV GPC into the GP2-293 packaging cell line (Clontech). A total of 5 × 106 GP2-293 cells were seeded on 10-cm plates and transfected 24 h later with 10 µg of DNA (5 µg of spike encoding plasmid and 5 µg of luciferase-encoding plasmid) using Lipofectamine 2000 (Invitrogen). Medium was replaced 5 h later with DMEM (Biological Industries) supplemented with 1% Pen-Strep (v/v), 1% glutamine (v/v), and 1% sodium pyruvate (v/v) and 10% fetal calf serum (FCS). At 48 h post-transfection, media containing pseudotyped viruses were collected, and VLPs were concentrated 10 times by the addition of PBS/8 % (w/v) PEG 6000 (Sigma), incubation at 4 °C for 24 h, centrifugation at 10,000g for 20 min and resuspension in full medium for viral depletion experiments or in plain DMEM for matriglycan inhibition assays. Concentrated VLPs were stored at −80 °C until use.
Generation of synthetic matriglycan in vitro
The LARGE1 (xylosyl- and glucuronyl-transferase I) gene (Forchheimer plasmid bank, Weizmann Institute of Science, Clone ID 100000367), without its cytosolic or transmembrane domains (residues 28–756) and with a 6× His tag at its N terminus, in pENTR223, was subcloned into a modified pHLsec downstream to a signal peptide, via its BglII and NotI restriction sites. Expression of LARGE1 was carried out in HEK 293F cells (Invitrogen) using FreeStyle Medium (Life Technologies). Cells were grown to a density of about 1.0 × 106 cells per ml before transfection. HEK 293F cells were transfected using 40 kDa polyethylenamine (PEI-MAX) (Polysciences) at 1 mg ml−1, pH 7 with DNA at a ratio of 1:2.5 (DNA:PEI solution). Soluble LARGE1 was collected from cell medium one week post-transfection. Cells were removed by centrifuged at 600g for 10 min and then the supernatant was spun down at 15,800g for 30 min to remove residual cellular debris. The supernatant was then supplemented with 100 μM PMSF and 0.02% sodium azide and filtered through a 0.45 μm Stericup (Merck Millipore). Buffer exchange to TBS (20 mM Tris-HCl, pH 8, 150 mM NaCl) was carried out using a Pellicon Tangential Flow Filtration system (Merck Millipore). LARGE1 was then captured using affinity chromatography on a 5 ml HiTrap IMAC FF Ni2+ column (GE Healthcare), and further purified using size-exclusion chromatography on a Superdex 200 10/300 GL column (GE Healthcare). Purified LARGE1 was concentrated using a 4 ml 30 kDa Amicon centrifugal unit (Sigma) and aliquots were flash frozen and stored at −80 °C.
Matriglycan was prepared in a reaction mixture containing 100 mM MES pH 5.8, 3.3 mM MgCl2, 3.3 mM MnCl2, 200 μM UDP-glucuronic acid (Promega), 200 μM UDP-xylose (CarboSource Services), 1.5 mM 4-nitrophenyl-α-d-xylopyranoside (used as a substrate for matriglycan elongation; Megazyme), and about 100 μg ml−1 of purified LARGE1. The reaction mixture was incubated at 37 °C for 60 h before flash freezing in liquid N2. To monitor matriglycan synthesis we used Promega’s glycosyltransferase kit. Specifically, 25 μl of a reaction mixture was mixed with 25 μl of UDP detection reagent and incubated for 1 h at room temperature. A calibration curve was derived using free UDP. Free UDP was then quantified by detecting bioluminescence signal following the addition of a UDP-Glo enzyme (Promega).
Inhibition of infectivity by matriglycan
For infectivity assays, HEK 293T cells (for LASV) or HEK 293T cells that overexpress human transferrin receptor60 (for MACV) were seeded on a poly-l-lysine-precoated white, chimney 96-well plate (Greiner Bio-One) at 50,000 cells per well. Cells were left to adhere for 3 h, followed by the addition of the supernatant containing the VLPs either with matriglycan (0.3 nmol of polymerized matriglycan units) or with matriglycan-generation buffer only as a control. Cells were washed from the viruses at 2 h post-infection, and luminescence from the activity of luciferase was measured at 48 h post-infection using Tecan Infinite M200 Pro plate reader after applying Bright-Glo reagent (Promega) to cells.
Depletion of pseudotyped viruses by IIH6
Protein-L beads (SCBT) were either loaded with IIH6 (40 µl of beads incubated with 150 µl of IIH6 at a concentration of 125 µg ml–1 for 30 min at room temperature), or washed using full medium (see above). Excess IIH6 was washed with 200 µl of full medium after centrifugation of the beads at 1,000g for 2 min, followed by 30 min incubation with 125 µl of either LASV or LUJV VLPs at room temperature. The supernatants containing the VLPs were separated using centrifugation at 1,000g for 2 min and transferred to a new tube for quantification by quantitative PCR with reverse transcription (RT–qPCR).
Quantifying pseudotyped viruses with RT–qPCR
A sample of 5 µl from previously described pulled-down pseudo-typed viruses was used to quantify RNA levels of the luciferase reporter gene using RT–qPCR. Viruses were first diluted 10× in PBS buffer, and clarified by centrifugation at 19,000g at 4 °C for 10 min. Supernatants were then treated with 2 mg ml−1 RNAse A (Bio Basic) and RNAse-free DNAse I (NEB) for 10 min at 37 °C to eliminate nucleic acids contaminations that are not protected inside the VLPs. To inhibit RNAse A activity, 40 units of RNase inhibitor (NEB) were added, and the reaction was incubated for 10 min at 37 °C. RNA was then extracted from viral-particles using the RNAeasy mini kit (Qiagen). Thereafter, cDNA was prepared from RNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). cDNA was diluted 1:40, and subjected to quantitative PCR (qPCR) using specific primers for luciferase, and Fast SYBR green master mix (Applied Biosystems).
Creating amino acid frequency plot
The amino acid frequency plot was generated using WebLogo61.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Coordinate files and experimental density maps for the C3-symmetric and C1-symmetric reconstructions were deposited at PDB (Electron Microscopy Data Bank (EMDB)) under accession codes 7PUY (EMD-13662) and 7PVD (EMD-13667), respectively. The C3-focused map was deposited to EMDB under accession code (EMD-13668).
Code availability
The complete script that was used to execute the Rosetta calculation is included in Supplementary Data.
References
McCormick, J. B., Webb, P. A., Krebs, J. W., Johnson, K. M. & Smith, E. S. A prospective study of the epidemiology and ecology of Lassa fever. J. Infect. Dis. 155, 437–444 (1987).
Bell-Kareem, A. R. & Smither, A. R. Epidemiology of Lassa fever. Cur. Top. Microbiol. Immunol. https://doi.org/10.1007/82_20 (2021).
Cao, W. et al. Identification of α-dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science 282, 2079–2081 (1998).
Kunz, S., Rojek, J. M., Perez, M., Spiropoulou, C. F. & Oldstone, M. B. Characterization of the interaction of Lassa fever virus with its cellular receptor α-dystroglycan. J. Virol. 79, 5979–5987 (2005).
Bederka, L. H., Bonhomme, C. J., Ling, E. L. & Buchmeier, M. J. Arenavirus stable signal peptide is the keystone subunit for glycoprotein complex organization. mBio 5, e02063 (2014).
Eichler, R. et al. Identification of Lassa virus glycoprotein signal peptide as a trans-acting maturation factor. EMBO Rep. 4, 1084–1088 (2003).
York, J., Romanowski, V., Lu, M. & Nunberg, J. H. The signal peptide of the Junin arenavirus envelope glycoprotein is myristoylated and forms an essential subunit of the mature G1–G2 complex. J. Virol. 78, 10783–10792 (2004).
York, J. & Nunberg, J. H. Role of the stable signal peptide of Junin arenavirus envelope glycoprotein in pH-dependent membrane fusion. J. Virol. 80, 7775–7780 (2006).
Agnihothram, S. S., York, J., Trahey, M. & Nunberg, J. H. Bitopic membrane topology of the stable signal peptide in the tripartite Junin virus GP-C envelope glycoprotein complex. J. Virol. 81, 4331–4337 (2007).
Froeschke, M., Basler, M., Groettrup, M. & Dobberstein, B. Long-lived signal peptide of lymphocytic choriomeningitis virus glycoprotein pGP-C. J. Biol. Chem. 278, 41914–41920 (2003).
Eichler, R. et al. Lassa virus glycoprotein signal peptide displays a novel topology with an extended endoplasmic reticulum luminal region. J. Biol. Chem. 279, 12293–12299 (2004).
Salam, A. P. et al. Time to reconsider the role of ribavirin in Lassa fever. PLoS Negl. Trop. Dis. 15, e0009522 (2021).
Radoshitzky, S. R. et al. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature 446, 92–96 (2007).
Abraham, J. et al. Host-species transferrin receptor 1 orthologs are cellular receptors for nonpathogenic new world clade B arenaviruses. PLoS Pathog. 5, e1000358 (2009).
Raaben, M. et al. NRP2 and CD63 are host factors for Lujo virus cell entry. Cell Host Microbe 22, 688–696 e685 (2017).
Yoshida-Moriguchi, T. & Campbell, K. P. Matriglycan: a novel polysaccharide that links dystroglycan to the basement membrane. Glycobiology 25, 702–713 (2015).
Kunz, S. et al. Posttranslational modification of alpha-dystroglycan, the cellular receptor for arenaviruses, by the glycosyltransferase LARGE is critical for virus binding. J. Virol. 79, 14282–14296 (2005).
Inamori, K. et al. Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science 335, 93–96 (2012).
Abraham, J., Corbett, K. D., Farzan, M., Choe, H. & Harrison, S. C. Structural basis for receptor recognition by New World hemorrhagic fever arenaviruses. Nat. Struct. Mol. Biol. 17, 438–444 (2010).
Cohen-Dvashi, H., Kilimnik, I. & Diskin, R. Structural basis for receptor recognition by Lujo virus. Nat. Microbiol. 3, 1153–1160 (2018).
Hastie, K. M. et al. Structural basis for antibody-mediated neutralization of Lassa virus. Science 356, 923–928 (2017).
Sullivan, B. M. et al. Point mutation in the glycoprotein of lymphocytic choriomeningitis virus is necessary for receptor binding, dendritic cell infection, and long-term persistence. Proc. Natl Acad. Sci. USA 108, 2969–2974 (2011).
Smelt, S. C. et al. Differences in affinity of binding of lymphocytic choriomeningitis virus strains to the cellular receptor α-dystroglycan correlate with viral tropism and disease kinetics. J. Virol. 75, 448–457 (2001).
Hastie, K. M. et al. Crystal structure of the prefusion surface glycoprotein of the prototypic arenavirus LCMV. Nat. Struct. Mol. Biol. 23, 513–521 (2016).
Cohen-Dvashi, H., Cohen, N., Israeli, H. & Diskin, R. Molecular mechanism for LAMP1 recognition by Lassa virus. J. Virol. 89, 7584–7592 (2015).
Israeli, H., Cohen-Dvashi, H., Shulman, A., Shimon, A. & Diskin, R. Mapping of the Lassa virus LAMP1 binding site reveals unique determinants not shared by other old world arenaviruses. PLoS Pathog. 13, e1006337 (2017).
Weinstein, J. Y., Elazar, A. & Fleishman, S. J. A lipophilicity-based energy function for membrane-protein modelling and design. PLoS Comput. Biol. 15, e1007318 (2019).
Saunders, A. A. et al. Mapping the landscape of the lymphocytic choriomeningitis virus stable signal peptide reveals novel functional domains. J. Virol. 81, 5649–5657 (2007).
Schrempf, S., Froeschke, M., Giroglou, T., von Laer, D. & Dobberstein, B. Signal peptide requirements for lymphocytic choriomeningitis virus glycoprotein C maturation and virus infectivity. J. Virol. 81, 12515–12524 (2007).
Briknarova, K., Thomas, C. J., York, J. & Nunberg, J. H. Structure of a zinc-binding domain in the Junin virus envelope glycoprotein. J. Biol. Chem. 286, 1528–1536 (2011).
Messina, E. L., York, J. & Nunberg, J. H. Dissection of the role of the stable signal peptide of the arenavirus envelope glycoprotein in membrane fusion. J. Virol. 86, 6138–6145 (2012).
Heijne, G. The distribution of positively charged residues in bacterial inner membrane proteins correlates with the trans-membrane topology. EMBO J. 5, 3021–3027 (1986).
Burri, D. J. et al. Differential recognition of Old World and New World arenavirus envelope glycoproteins by subtilisin kexin isozyme 1 (SKI-1)/site 1 protease (S1P). J. Virol. 87, 6406–6414 (2013).
Ervasti, J. M. & Campbell, K. P. Membrane organization of the dystrophin–glycoprotein complex. Cell 66, 1121–1131 (1991).
Acciani, M. et al. Mutational analysis of Lassa virus glycoprotein highlights regions required for α-dystroglycan utilization. J. Virol. 91, e00574–17 (2017).
Prod’homme, V. et al. Human cytomegalovirus UL40 signal peptide regulates cell surface expression of the NK cell ligands HLA-E and gpUL18. J. Immunol. 188, 2794–2804 (2012).
Cocquerel, L. et al. Topological changes in the transmembrane domains of hepatitis C virus envelope glycoproteins. EMBO J. 21, 2893–2902 (2002).
Cohen-Dvashi, H., Israeli, H., Shani, O., Katz, A. & Diskin, R. Role of LAMP1 binding and pH sensing by the spike complex of Lassa virus. J. Virol. 90, 10329–10338 (2016).
Bigman, L. S., Greenblatt, H. M. & Levy, Y. What are the molecular requirements for protein sliding along DNA? J. Phys. Chem. B 125, 3119–3131 (2021).
Johnson, B. A. et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature 591, 293–299 (2021).
Cantuti-Castelvetri, L. et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 370, 856–860 (2020).
Smith, T. J. et al. The site of attachment in human rhinovirus 14 for antiviral agents that inhibit uncoating. Science 233, 1286–1293 (1986).
Rossmann, M. G. et al. Structure of a human common cold virus and functional relationship to other picornaviruses. Nature 317, 145–153 (1985).
Toelzer, C. et al. Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science 370, 725–730 (2020).
Pullikotil, P., Benjannet, S., Mayne, J. & Seidah, N. G. The proprotein convertase SKI-1/S1P: alternate translation and subcellular localization. J. Biol. Chem. 282, 27402–27413 (2007).
Burri, D. J. et al. Molecular characterization of the processing of arenavirus envelope glycoprotein precursors by subtilisin kexin isozyme-1/site-1 protease. J. Virol. 86, 4935–4946 (2012).
Grewal, P. K., McLaughlan, J. M., Moore, C. J., Browning, C. A. & Hewitt, J. E. Characterization of the LARGE family of putative glycosyltransferases associated with dystroglycanopathies. Glycobiology 15, 912–923 (2005).
Willer, T. et al. The glucuronyltransferase B4GAT1 is required for initiation of LARGE-mediated alpha-dystroglycan functional glycosylation. eLife 3, e03941 (2014).
Osman Sheikh, M. et al. Cell surface glycan engineering reveals that matriglycan alone can recapitulate dystroglycan binding and function. Preprint at bioRxiv https://doi.org/10.1101/2021.05.10.443358 (2021).
Paroutis, P., Touret, N. & Grinstein, S. The pH of the secretory pathway: measurement, determinants, and regulation. Physiology 19, 207–215 (2004).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010).
The PyMOL Molecular Graphics System. Version 1.8 (2016).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D 67, 235–242 (2011).
Lomize, M. A., Pogozheva, I. D., Joo, H., Mosberg, H. I. & Lomize, A. L. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 40, D370–D376 (2012).
Elazar, A. et al. Mutational scanning reveals the determinants of protein insertion and association energetics in the plasma membrane. eLife 5, e12125 (2016).
Fleishman, S. J. et al. RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. PLoS ONE 6, e20161 (2011).
Cohen-Dvashi, H. et al. Rational design of universal immunotherapy for TfR1-tropic arenaviruses. Nat. Commun. 11, 67 (2020).
Crooks, G. E., Hon, G., Chandonia, J. M. & Brenner, S. E. WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 (2004).
Acknowledgements
The Diskin laboratory is supported by research grants from the Ernst I. Ascher foundation, Ben B. and Joyce E. Eisenberg Foundation, Estate of Emile Mimran, Jeanne and Joseph Nissim Center for Life Sciences Research, Dov and Ziva Rabinovich Endowed Fund for Structural Biology, Donald Rivin, Stanley and Tanya Rossby Endowment Fund, Natan Sharansky, Dr. Barry Sherman Institute for Medicinal Chemistry, as well as from the Israel Science Foundation (grants No. 3147/19 and 209/20). Research in the Fleishman laboratory was supported by a Consolidator Award from the European Research Council (815379) and by a charitable donation in memory of Sam Switzer.
Author information
Authors and Affiliations
Contributions
R.D. conceived this research. M.K. and K.G. produced and purified the protein. M.K., N.E. and R.D. collected electron microscopy data. M.K. and R.D. solved the structure. J.W. and S.J.F. computationally determined the SSP register. H.C.-D. performed qPCR experiments and provided reagents. M.E.-A. performed infectivity assays. All authors contributed to data analysis. M.K. and R.D. prepared the manuscript with help from all the other authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Juha Huiskonen, Félix Rey and Yi Shi for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Reconstruction of density map.
The course of data processing and reconstruction of a C3-symmetric density map is visually summarized. Local resolution estimates, gold-standard FSC curve and orientational distribution are shown for the final map.
Extended Data Fig. 2 Overall fit of the model to the density map.
Model is shown as a ribbon with or without stick-represented side chains. Density map is shown using a blue mesh at the indicated sigma levels. Asterisks indicate that the density map was carved around the model. Upper left side, the original model was fitted into a working map at GSFSC = 2.5 Å and achieved map-model FSC of 3.3 Å. Center, final refined model at a 3.3 Å low-passed filtered map with a map-model FSC of 3.3 Å. Around the central model, close-up views of regions of interest are indicated at the 3.3 Å low-passed filtered map. Key residues or structural elements are labeled. Bottom part, the six-helical transmembrane bundle is shown in a 6 Å low-pass filtered map at the indicated three different sigma levels.
Extended Data Fig. 3 Assigning the overall directionality and registry of the SSP.
a. The amino-terminus of the SSP’s trans-membrane α-helix is facing extracellularly. The density map of the trans-membrane α-helix is shown as a mesh, and two α-helices are rendered as poly-ala; one has a carboxy-terminus facing up (left) and the second has an amino-terminus facing up (right). Densities for the side-chains are pointing toward the extracellular direction (up), indicating that the correct directionality of this α-helix in the membrane is with its amino-terminus at the extracellular region (right). b. Side-chain densities are visible in low-pass filtered maps. Densities for the α-helix of SSP are shown with maps that were low-pass (LP) filtered to the indicated resolution values. Dashed horizontal lines help to visualize the direction to which the side-chains are pointing. The directionality of the side-chains is apparent even at 4.5 Å LP-filtered map. c. Determining the registry of the SSP. On the left, superimposition of the 37 best-scoring computer-generated models for each threading (N-terminus out, only), shown in a ‘top’ view. The inner three α-helices (yellow, brown, and gray) belong to the GP2 subunits and the outer three α-helices (magenta, cyan, and green) belong to the SSP subunits. On the right, energy profiles (relative Rosetta energy units) of the best-scoring computed model for each threading option as a function of the register, starting with the indicated residue at the amino-terminus of the trans-membrane α-helix (first methionine is considered as register “0”). The light-blue curve shows threading with the C-terminus out and the red curve shows threading with the N-terminus out. Large red dot indicates the lowest energy solution. d. The top-ranked model for the SSP registry.
Extended Data Fig. 4 Domain swapping stabilization of the spike in a double-sided candy twist-wrap fashion.
The directionality of the domain swapping in the trimer is illustrated. The domain swapping at the apex of the trimer (top) and at the membrane proximal region (bottom) have opposing directionalities, which is reminiscence to a candy’s double-sided twist wrap (schematically shown on the right). Donation of a structural element from one subunit to another is marked by ‘–****;’.
Extended Data Fig. 5 Reconstruction of focused density maps.
The process of reconstructing focused density maps is visually illustrated.
Extended Data Fig. 6 Matriglycan on pseudotyped viruses and in density map.
a. Free Flag peptide is not recognized by anti-Flag antibody in a dot-blot settings. To test if the anti-Flag antibody could recognize the Flag peptide that was used to elute the spike complexes, we spotted on a nitrocellulose membrane the LASV and MACV spike complexes as well as the elution buffer alone, or the elution buffer with the Flag peptide (FLAG 1x). The elution buffer with the free Flag peptide produces only slight background, indicating that the Flag peptide is not adhering to the membrane, and hence does not contribute to the signal of the anti-Flag antibody. b. LASV pseudotyped viruses are depleted from solution by IIH6 conjugated to beads. To test if LASV spike complex is loaded with matriglycan in the context of pseudotyped viruses, we incubated MLV-based pseudotyped viruses bearing the spikes of LASV or of the neuropilin-2-tropic Lujo virus (LUJV) with protein-L beads alone, or after coating the beads with the IIH6 antibody. The amount of RNA encoding a luciferase reporter gene was then quantified in the supernatants of each sample using RT-qPCR. Each dot represents an average normalized value of three technical replicates in a single independent experiment (n = 5 of independent experiments). The measurements in each experiment were normalized to the respective uncoated-beads control. Pseudotyped viruses with the LASV spike are significantly (two tailed Student’s t-test) depleted from solution by IIH6 on beads. Whiskers indicate the min and max values, central line indicates the mean value, and the box indicates the interquartile range. The slight increase in the amount of LUJV results from minor unspecific absorption of viral particles to uncoated protein-L beads, which lowers the amount of particle in the reference control. c. The ectodomain portion of the spike was fitted into the C1-focused map and yielded a map-model FSC of 3.7 Å. The map was low-passed filtered to 3.7 Å and the model was refined again using this map. The linear matriglycan chain is shown by its top and side views. The density map is shown as a blue mesh at σ = 4, carved at 2.5 Å around the matriglycan. The Xyl-GlcA-Xyl moieties in the symmetric sites are in orange. The GlcA moieties in the asymmetric sites as well as the termini Xyl-GlcA are shown in green.
Extended Data Fig. 7 Residues that affect matriglycan binding and a comparison of the cryo-EM structure of LASV spike with the crystal structure of its ectodomain.
a. His141 and Phe147 (blue) that were found to be important for matriglycan binding via mutagenesis studies are located directly below the “RRLL” motif (magenta) that binds matriglycan (orange). The conformation of the “RRLL” motif is partially shaped by the interactions it makes with His141 and Phe147. b. The cryo-EM model from this study (similar color scheme as in Fig. 1) is show with the crystal structure of the spike’s ectodomain (PDB: 5VK2, green). The two structures are shown as ribbons from a side view (left) and from a top view (top right). Overall, the structures are similar (RMSD = 2.04 Å, for 1009 shared Cα atoms). Key differences are near the membrane spanning domain that is missing from the crystal structure and at the receptor binding site that is not formed (enlarged view, bottom right), following the replacement of the “RRLL”-SKI-I recognition site with a furin recognition sequence. Tyr150, Arg256, and Arg257 that participate in binding of matriglycan are labeled.
Extended Data Fig. 8 Degenerate and alternative states for matriglycan binding.
A top view of the LASV trimeric spike showing a surface representation of the three GP1 subunits in pink, blue, and gray. Colored dots represent the potential traces of bound matriglycan chains. Different colors represent different individual chains of matriglycan.
Extended Data Fig. 9 Cleavage sites between the receptor binding modules and the transmembrane modules in different viruses.
The trimeric spike complexes of LASV (left) and of SARS-CoV-2 (right, PDB: 6VXX), are shown in a relative scale using surface representations and in three different colors for their three protomers. The protease cleavage sites (i.e., a SKI-I site for LASV and a furin site for SARS-CoV-2) or their immediate vicinities are colored in orange and are pointed with red arrows.
Supplementary information
Supplementary Information
This file contains the Supplementary Methods, which includes the command line and script for Rosetta modelling.
Rights and permissions
About this article

Cite this article
Katz, M., Weinstein, J., Eilon-Ashkenazy, M. et al. Structure and receptor recognition by the Lassa virus spike complex. Nature 603, 174–179 (2022). https://doi.org/10.1038/s41586-022-04429-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-04429-2
This article is cited by
-
Cleavage-intermediate Lassa virus trimer elicits neutralizing responses, identifies neutralizing nanobodies, and reveals an apex-situated site-of-vulnerability
Nature Communications (2024)
-
Cell surface glycan engineering reveals that matriglycan alone can recapitulate dystroglycan binding and function
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
