
Abstract
Identifying the relationships between chromosome structures, nuclear bodies, chromatin states and gene expression is an overarching goal of nuclear-organization studies1,2,3,4. Because individual cells appear to be highly variable at all these levels5, it is essential to map different modalities in the same cells. Here we report the imaging of 3,660 chromosomal loci in single mouse embryonic stem (ES) cells using DNA seqFISH+, along with 17 chromatin marks and subnuclear structures by sequential immunofluorescence and the expression profile of 70 RNAs. Many loci were invariably associated with immunofluorescence marks in single mouse ES cells. These loci form ‘fixed points’ in the nuclear organizations of single cells and often appear on the surfaces of nuclear bodies and zones defined by combinatorial chromatin marks. Furthermore, highly expressed genes appear to be pre-positioned to active nuclear zones, independent of bursting dynamics in single cells. Our analysis also uncovered several distinct mouse ES cell subpopulations with characteristic combinatorial chromatin states. Using clonal analysis, we show that the global levels of some chromatin marks, such as H3 trimethylation at lysine 27 (H3K27me3) and macroH2A1 (mH2A1), are heritable over at least 3–4 generations, whereas other marks fluctuate on a faster time scale. This seqFISH+-based spatial multimodal approach can be used to explore nuclear organization and cell states in diverse biological systems.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Data availability
The source data and processed data from this study are available at Zenodo (https://zenodo.org/record/3735329). Additional raw microscopy data obtained during this study are available from the corresponding author upon reasonable request. Publicly available datasets used in the study (GSE96107, 4DNESOJRTZZR, GSE17051, GSE102076, GSE48895, ENCSR000CFN, ENCSR000CGP, ENCSR000CGQ) are detailed in the Methods.
Code availability
The custom written scripts used in this study are available at https://github.com/CaiGroup/dna-seqfish-plus.
References
Dekker, J. et al. The 4D nucleome project. Nature 549, 219–226 (2017).
Kelsey, G., Stegle, O. & Reik, W. Single-cell epigenomics: recording the past and predicting the future. Science 358, 69–75 (2017).
Kempfer, R. & Pombo, A. Methods for mapping 3D chromosome architecture. Nat. Rev. Genet. 21, 207–226 (2020).
Zhu, C., Preissl, S. & Ren, B. Single-cell multimodal omics: the power of many. Nat. Methods 17, 11–14 (2020).
Finn, E. H. & Misteli, T. Molecular basis and biological function of variability in spatial genome organization. Science 365, eaaw9498 (2019).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009).
Quinodoz, S. A. et al. Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell 174, 744–757 (2018).
Wang, S. et al. Spatial organization of chromatin domains and compartments in single chromosomes. Science 353, 598–602 (2016).
Bintu, B. et al. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362, eaau1783 (2018).
Nir, G. et al. Walking along chromosomes with super-resolution imaging, contact maps, and integrative modeling. PLoS Genet. 14, e1007872 (2018).
Cardozo Gizzi, A. M. et al. Microscopy-based chromosome conformation capture enables simultaneous visualization of genome organization and transcription in intact organisms. Mol. Cell 74, 212–222 (2019).
Finn, E. H. et al. Extensive heterogeneity and intrinsic variation in spatial genome organization. Cell 176, 1502–1515 (2019).
Mateo, L. J. et al. Visualizing DNA folding and RNA in embryos at single-cell resolution. Nature 568, 49–54 (2019).
Nguyen, H. Q. et al. 3D mapping and accelerated super-resolution imaging of the human genome using in situ sequencing. Nat. Methods 17, 822–832 (2020).
Su, J.-H., Zheng, P., Kinrot, S. S., Bintu, B. & Zhuang, X. Genome-scale imaging of the 3d organization and transcriptional activity of chromatin. Cell 182, 1641–1659 (2020).
Beliveau, B. J. et al. Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proc. Natl Acad. Sci. USA 109, 21301–21306 (2012).
Lubeck, E., Coskun, A. F., Zhiyentayev, T., Ahmad, M. & Cai, L. Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 11, 360–361 (2014).
Shah, S., Lubeck, E., Zhou, W. & Cai, L. In situ transcription profiling of single cells reveals spatial organization of cells in the mouse hippocampus. Neuron 92, 342–357 (2016).
Takei, Y., Shah, S., Harvey, S., Qi, L. S. & Cai, L. Multiplexed dynamic imaging of genomic loci by combined CRISPR imaging and DNA sequential FISH. Biophys. J. 112, 1773–1776 (2017).
Shah, S. et al. Dynamics and spatial genomics of the nascent transcriptome by intron seqFISH. Cell 174, 363–376 (2018).
Eng, C. L. et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature 568, 235–239 (2019).
Chen, K. H., Boettiger, A. N., Moffitt, J. R., Wang, S. & Zhuang, X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015).
Bonev, B. et al. Multiscale 3D genome rewiring during mouse neural development. Cell 171, 557–572.e24 (2017).
Shen, Y. et al. A map of the cis-regulatory sequences in the mouse genome. Nature 488, 116–120 (2012).
Boettiger, A. N. et al. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 529, 418–422 (2016).
van Steensel, B. & Belmont, A. S. Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 169, 780–791 (2017).
Spector, D. L. & Lamond, A. I. Nuclear speckles. Cold Spring Harb. Perspect. Biol. 3, a000646 (2011).
Pederson, T. The nucleolus. Cold Spring Harb. Perspect. Biol. 3, a000638 (2011).
Ludwig, C. H. & Bintu, L. Mapping chromatin modifications at the single cell level. Development 146, dev170217 (2019).
Söderberg, O. et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 (2006).
Agasti, S. S. et al. DNA-barcoded labeling probes for highly multiplexed Exchange-PAINT imaging. Chem. Sci. 8, 3080–3091 (2017).
Guenatri, M., Bailly, D., Maison, C. & Almouzni, G. Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J. Cell Biol. 166, 493–505 (2004).
Solovei, I. et al. Nuclear architecture of rod photoreceptor cells adapts to vision in mammalian evolution. Cell 137, 356–368 (2009).
Mao, Y. S., Zhang, B. & Spector, D. L. Biogenesis and function of nuclear bodies. Trends Genet. 27, 295–306 (2011).
Peric-Hupkes, D. et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol. Cell 38, 603–613 (2010).
Kind, J. et al. Genome-wide maps of nuclear lamina interactions in single human cells. Cell 163, 134–147 (2015).
Chen, Y. et al. Mapping 3D genome organization relative to nuclear compartments using TSA-seq as a cytological ruler. J. Cell Biol. 217, 4025–4048 (2018).
Gut, G., Herrmann, M. D. & Pelkmans, L. Multiplexed protein maps link subcellular organization to cellular states. Science 361, eaar7042 (2018).
McSwiggen, D. T., Mir, M., Darzacq, X. & Tjian, R. Evaluating phase separation in live cells: diagnosis, caveats, and functional consequences. Genes Dev. 33, 1619–1634 (2019).
Marks, H. et al. The transcriptional and epigenomic foundations of ground state pluripotency. Cell 149, 590–604 (2012).
Singer, Z. S. et al. Dynamic heterogeneity and DNA methylation in embryonic stem cells. Mol. Cell 55, 319–331 (2014).
Kolodziejczyk, A. A. et al. Single cell RNA-sequencing of pluripotent states unlocks modular transcriptional variation. Cell Stem Cell 17, 471–485 (2015).
Tosolini, M. et al. Contrasting epigenetic states of heterochromatin in the different types of mouse pluripotent stem cells. Sci. Rep. 8, 5776 (2018).
van Mierlo, G. et al. Integrative proteomic profiling reveals PRC2-dependent epigenetic crosstalk maintains ground-state pluripotency. Cell Stem Cell 24, 123–137.e8 (2019).
Gerlich, D. et al. Global chromosome positions are transmitted through mitosis in mammalian cells. Cell 112, 751–764 (2003).
Walter, J., Schermelleh, L., Cremer, M., Tashiro, S. & Cremer, T. Chromosome order in HeLa cells changes during mitosis and early G1, but is stably maintained during subsequent interphase stages. J. Cell Biol. 160, 685–697 (2003).
Thomson, I., Gilchrist, S., Bickmore, W. A. & Chubb, J. R. The radial positioning of chromatin is not inherited through mitosis but is established de novo in early G1. Curr. Biol. 14, 166–172 (2004).
Essers, J. et al. Dynamics of relative chromosome position during the cell cycle. Mol. Biol. Cell 16, 769–775 (2005).
Hormoz, S. et al. inferring cell-state transition dynamics from lineage trees and endpoint single-cell measurements. Cell Syst. 3, 419–433 (2016).
Cunningham, F. et al. Ensembl 2019. Nucleic Acids Res. 47, D745–D751 (2019).
Klein, A. M. et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 (2015).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 6, 11 (2015).
Eng, C. L., Shah, S., Thomassie, J. & Cai, L. Profiling the transcriptome with RNA SPOTs. Nat. Methods 14, 1153–1155 (2017).
Cremer, C. et al. in Handbook of Computer Vision and Applications Vol. 3 (ed. Jahne, B. et al) 839–857 (Academic, 1999).
Croft, J. A. et al. Differences in the localization and morphology of chromosomes in the human nucleus. J. Cell Biol. 145, 1119–1131 (1999).
Esa, A. et al. Three-dimensional spectral precision distance microscopy of chromatin nanostructures after triple-colour DNA labelling: a study of the BCR region on chromosome 22 and the Philadelphia chromosome. J. Microsc. 199, 96–105 (2000).
Cremer, M. et al. Multicolor 3D fluorescence in situ hybridization for imaging interphase chromosomes. Methods Mol. Biol. 463, 205–239 (2008).
Zhang, Z., Revyakin, A., Grimm, J. B., Lavis, L. D. & Tjian, R. Single-molecule tracking of the transcription cycle by sub-second RNA detection. eLife 3, e01775 (2014).
Chen, B. et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155, 1479–1491 (2013).
Nilsson, M. et al. Padlock probes: circularizing oligonucleotides for localized DNA detection. Science 265, 2085–2088 (1994).
Rouhanifard, S. H. et al. ClampFISH detects individual nucleic acid molecules using click chemistry-based amplification. Nat. Biotechnol. (2018). https://doi.org/10.1038/nbt.4286
Edelstein, A., Amodaj, N., Hoover, K., Vale, R. & Stuurman, N. Computer control of microscopes using μManager. Curr. Protoc. Mol. Biol. Ch. 14, Unit14.20 (2010).
Liu, S.-L. et al. Fast and high-accuracy localization for three-dimensional single-particle tracking. Sci. Rep. 3, 2462 (2013).
Parthasarathy, R. Rapid, accurate particle tracking by calculation of radial symmetry centers. Nat. Methods 9, 724–726 (2012).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Knight, P. A. & Ruiz, D. A fast algorithm for matrix balancing. IMA J. Numer. Anal. 33, 1029–1047 (2013).
Stanyte, R. et al. Dynamics of sister chromatid resolution during cell cycle progression. J. Cell Biol. 217, 1985–2004 (2018).
McInnes, L., Healy, J., Saul, N. & Großberger, L. UMAP: uniform manifold approximation and projection. J. Open Source Softw. 3, 861 (2018).
Nmezi, B. et al. Concentric organization of A- and B-type lamins predicts their distinct roles in the spatial organization and stability of the nuclear lamina. Proc. Natl Acad. Sci. USA 116, 4307–4315 (2019).
Suzuki, H., Kurihara, Y., Kanehisa, T. & Moriwaki, K. Variation in the distribution of silver-staining nucleolar organizer regions on the chromosomes of the wild mouse, Mus musculus. Mol. Biol. Evol. 7, 271–282 (1990).
Kurihara, Y., Suh, D. S., Suzuki, H. & Moriwaki, K. Chromosomal locations of Ag-NORs and clusters of ribosomal DNA in laboratory strains of mice. Mamm. Genome 5, 225–228 (1994).
Strongin, D. E., Groudine, M. & Politz, J. C. R. Nucleolar tethering mediates pairing between the IgH and Myc loci. Nucleus 5, 474–481 (2014).
Jonkers, I., Kwak, H. & Lis, J. T. Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. eLife 3, e02407 (2014).
Dileep, V. & Gilbert, D. M. Single-cell replication profiling to measure stochastic variation in mammalian replication timing. Nat. Commun. 9, 427 (2018).
Bacher, R. et al. SCnorm: robust normalization of single-cell RNA-seq data. Nat. Methods 14, 584–586 (2017).
Vallejos, C. A., Risso, D., Scialdone, A., Dudoit, S. & Marioni, J. C. Normalizing single-cell RNA sequencing data: challenges and opportunities. Nat. Methods 14, 565–571 (2017).
Hafemeister, C. & Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019).
Traag, V. A., Waltman, L. & van Eck, N. J. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 9, 5233 (2019).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
Serrano, L. et al. The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Genes Dev. 27, 639–653 (2013).
Hastie, T. & Stuetzle, W. Principal curves. J. Am. Stat. Assoc. 84, 502–516 (1989).
Haghverdi, L., Büttner, M., Wolf, F. A., Buettner, F. & Theis, F. J. Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods 13, 845–848 (2016).
van Dijk, D. et al. Recovering gene interactions from single-cell data using data diffusion. Cell 174, 716–729 (2018).
Angerer, P. et al. destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics 32, 1241–1243 (2016).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Lyon, M. F. X-chromosome inactivation: a repeat hypothesis. Cytogenet. Cell Genet. 80, 133–137 (1998).
Meuleman, W. et al. Constitutive nuclear lamina-genome interactions are highly conserved and associated with A/T-rich sequence. Genome Res. 23, 270–280 (2013).
Acknowledgements
We thank I. Strazhnik for help with figures; A. Anderson for help with the manuscript; C. Karp for custom-made flow cells; H. -J. Ahn for the early phase of the antibody conjugation; and B. Bonev for the Hi-C data. This project is funded by NIH 4DN DA047732 and supplement, and the Paul G. Allen Frontiers Foundation Discovery Center.
Author information
Authors and Affiliations
Contributions
Y.T. and L.C. conceived the idea and designed experiments. Y.T. designed probes with help from J.T. and C.-H.L.E. Y.T. and J.Y. prepared and validated all the experimental materials. Y.T. performed all the experiments with help from J.Y. Y.T. and N.P. performed image analysis with help from J.W. and S. Shah. Y.T., S.Z. and L.C. analysed data with N.O. and S. Suo. L.C., M.G. and G.-C.Y. supervised the analysis process. Y.T. and L.C. wrote the manuscript with input from C.-H.L.E. and G.-C.Y. L.C. supervised all aspects of the projects.
Corresponding author
Ethics declarations
Competing interests
L.C. is a co-founder of Spatial Genomics Inc.
Additional information
Peer review information Nature thanks Peter Fraser and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
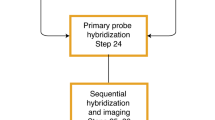
Extended Data Fig. 1 Detailed schematics of the integrated spatial genomics approach with DNA seqFISH+, RNA and intron seqFISH and multiplexed immunofluorescence.
a, Flow chart of the experimental procedures. Samples are fixed with paraformaldehyde, followed by oligonucleotide-conjugated primary antibody incubation, post-fixation with paraformaldehyde and BS(PEG)5, and RNA seqFISH. Then samples are prepared for DNA seqFISH+. This optimized protocol ensures good alignment between DNA seqFISH+ data with RNA seqFISH and the multiplexed immunofluorescence data on a voxel by voxel level (Extended Data Fig. 2). Bottom right cartoon shows imaging routine for RNA FISH and DNA seqFISH+ with primary probes and sequential immunofluorescence with oligonucleotide-conjugated primary antibodies. b, Schematics of DNA seqFISH+ for the 1-Mb resolution dataset. Five rounds of barcoding allows 2,048 barcodes to be detected with 2 rounds of dropout error correction in each fluorescent channel. Two fluorescent channels are used to cover a total of 2,460 loci, spaced approximately 1 Mb apart in the genome. In each round of barcoding, 16 rounds of hybridization are performed to generate 16 pseudocolors. DNA dots detected in each pseudocolor channel are fitted in 3D to determine their super-resolved centroid location and compiled across all 16 pseudocolors to generate a super-resolved localization image. With 5 rounds of barcoding (overall 80 rounds of serial hybridizations), the identity of all DNA loci are decoded. Every DNA locus should appear once in every barcoding round in a single pseudocolor. The barcoding table (Supplementary Table 2) is shown on the right. DNA seqFISH+ probes contain all 5 rounds of barcode readout sequences. Each sequence, for a given barcoding round, has a possible choice of 16 sequences, corresponding to one of the pseudocolors. For each gene, 5 out of the 80 hybridizations will result in hybridization events and fluorescent readout probes bound on the primary DNA hybridizing probes. To preserve the DNA primary probe on the chromosome over all 80 rounds of hybridizations, the primary probes are padlocked62,63 onto the chromosomes by T4 DNA ligase at the primer binding sites after the initial hybridization (Methods). c, Barcode scheme for the 25-kb resolution DNA seqFISH+. 60 adjacent 25-kb regions are sequentially readout and imaged in 60 rounds of hybridization. This is carried out in parallel on 20 chromosomes. In other words, each round of hybridization images 20 different loci on different chromosomes. An additional 20 rounds of hybridization are carried out to label each chromosome one at a time to assign chromosomal identity to each locus imaged during the first 60 rounds individually. The 1-Mb resolution data were collected in the 643-nm (channel 1) and 561-nm (channel 2) channels in b, while the 25 kb resolution data were collected in the 488-nm channel (channel 3) in c.
Extended Data Fig. 2 Optimization and validation for DNA seqFISH+.
a, Ligation and post-fixation of primary probes prevent their dissociation at the readout probe stripping step, validated by telomere DNA FISH. 55% formamide wash buffer (WB) solution at 37 °C was added to the cells for 16 h with and without the primary probes padlocked62,63 onto the chromosomal DNA. Probes were retained in the ligated sample, and not retained in the unligated sample. Note that 55% WB was used at room temperature for 2 min in each stripping step during the seqFISH routine, which is less stringent than the condition used here. b, Quantification of the signal retention after the harsh wash in a, with telomere DNA FISH across multiple conditions. Total intensities in individual nuclei from a single z-section were compared before and after the harsh wash. In the DNA seqFISH+ experiments, the condition with ligation and post-fixation was used. The number of cells from two independent measurements is written in the plot. For the box plots in b and g, the centre line in the boxes marks median, the upper and lower limits of the boxes mark the interquartile range, the whiskers extend to the farthest data points within 1.5 times the interquartile range, and the grey points mark outliers. c, Primary probes are still bound after more than 81 rounds of hybridization, and the specific signals return in the DNA seqFISH+ experiments. Initial hyb0 for DNA seqFISH+ was performed with hyb80 readout probes for comparison. Fiducial markers targeting a repetitive region of the genome with a single primary probe were also imaged initially and included in all 80 imaging rounds for alignment. d, Quantification of the fiducial marker intensities for 80 hybridization rounds in the DNA seqFISH+ experiments, relative to that from hyb0 fiducial markers. Fiducial markers (n = 506-1,117 dots per hybridization round) from 446 cells in DNA seqFISH+ experiments were used for quantification. Shaded regions represent the mean (centre) with s.d. e, Localization errors of fiducial markers across hyb 1 to 80 in the DNA seqFISH+ experiments, n = 71,981 aligned spots for x and y, and n = 87,879 aligned spots for z from 446 cells in DNA seqFISH+ experiments. For x and y alignments, we filtered out aligned dots that were more than 2× s.d. away from the mean displacement at each hybridization, and new alignments were computed. f, Preservation of the nuclear structure through the double fixation procedure. Good colocalization (yellow in the right panel) of the nuclear speckles (SF3A66) before and after heating. g, Quantification of the SF3A66 immunofluorescence signal retention in the nuclei (left) and localization precision (right) measured by Pearson correlation of pixel intensities in the nuclei with a single z-section between hyb0 (pre-DNA seqFISH+ steps) image and hyb40 (pre-DNA seqFISH+ steps) or hyb130 (post-DNA seqFISH+ steps). n = 326 cells in the centre field of views from two DNA seqFISH+ biological replicates in g–k. h, Frequencies of on- and off-target barcodes in channel 1 and 2 per cell. On average, 3,636.0 ± 1,052.6 (median ± s.d.) on-target barcodes and 14.0 ± 7.4 off-target barcodes are detected per cell (n = 326 cells from the centre field of views of the two biological replicates). i, Average frequencies of individual on-target and off-target barcodes (n = 4,096 barcodes in channel 1 and 2), demonstrating the accuracy of the DNA seqFISH+. j, The total number of dots detected in each of the fluorescent channels in single cells. Channels 1 and 2 contain the 1-Mb data and channel 3 contains the 25-kb data. k, The average number of dots detected per each locus per cell across all 20 chromosomes. Note that 2 dots per cell are not 100% detection efficiency because some cells are in the G2 phase of the cell cycle (4 alleles in total). X chromosome has half the number of dots detected per locus (0.84 ± 0.21 (median ± s.d.)) compared with the other autosomes (1.57 ± 0.27), because E14 mouse ES cells are a male diploid cell line (Methods). l, Pearson correlation of probabilities for the pairs of loci within a search radius of 500 nm (1-Mb data) and 150 nm (25-kb data) between two biological replicates of DNA seqFISH+ experiments. All unique intra-chromosomal pairs of loci were calculated for the 1-Mb (n = 2,460 loci) and 25-kb data (n = 1,200 loci) with n = 201, 245 cells for biological replicates 1 and 2, respectively. m, PCC of the proximity probability between loci pairs as a function of search radii in comparison to 500-nm search radius (1-Mb data) and 150-nm search radius (25-kb data) used in l. n = 446 cells from the two DNA seqFISH+ biological replicates.
Extended Data Fig. 3 Additional validation for DNA seqFISH+.
a, b, Spearman correlation between probabilities of pairs of loci within a search radius of 100 nm–2 μm by DNA seqFISH+ and frequencies by Hi-C23 in mouse ES cells with a certain bin size. All unique intra-chromosomal pairs of loci were calculated for the 1-Mb (n = 2,340 autosomal loci) and 25-kb data (n = 60 loci per chromosome), and overlapping regions within the bin in a were excluded from this analysis. At 1.5 Mb chromosomal regions with 25 kb resolution in b, median Hi-C reads vary depending on the 1.5 Mb regions targeted, ranging from 0.9 to 203.2. We used 5 autosomal regions with Hi-C reads greater than 40 per 25-kb bin for comparison. c, Comparison of probabilities within 500-nm search radius for intra-chromosomal locus pairs in autosomes in DNA seqFISH+ (1-Mb resolution data) and the frequencies in Hi-C23 data in mouse ES cells. Spearman correlation coefficient of 0.89 computed from n = 84,707 unique intra-chromosomal pairwise combinations. Hi-C data were binned with 1-Mb data, and overlapping regions within 1 Mb were excluded from this analysis. d, Comparison of probabilities within a 500-nm search radius for the intra-chromosomal locus pairs in autosomes by DNA seqFISH+ (1-Mb resolution data) and frequencies by SPRITE7 in mouse ES cells. Spearman correlation coefficient of 0.83. The same binning and filtering were used as the Hi-C analysis in c. e, Comparison of probabilities within 150-nm search radius for the locus pairs in the selected autosomes by DNA seqFISH+ (25-kb resolution data) and frequences by Hi-C23 in mouse ES cells. Spearman correlation coefficients ranged from 0.82 to 0.94 computed from n = 948–1,776 unique pairwise combinations, using the same selection and filtering criteria as b. f, g, Relationships between median spatial distance of pairs of loci for 1-Mb resolution data in f and 25-kb resolution data in g by DNA seqFISH+ and Hi-C frequencies. The red lines are power-law fits with fitting parameters S shown with Spearman correlation coefficient R. h, i, Heat maps showing probabilities of pairs of loci within a search radius of 500 nm in h and 150 nm in i (top right triangles), and median spatial distances of pairs of loci (bottom left triangles) in each chromosome for 1 Mb resolution data in h and 25-kb resolution data in i by DNA seqFISH+. n = 446 cells from two biological replicates for DNA seqFISH+ data in a–i.
Extended Data Fig. 4 Single-cell organization and physical scaling of chromosomes by DNA seqFISH+.
a, DAPI staining image of mouse ES cells (top) and 3D image of corresponding nuclei with individual chromosomes labelled with different colours (bottom). b, 3D image of individual chromosomes, coloured based on chromosome coordinates (light to dark colours). Chromosomes are from cells in a. The images are representative of n = 446 cells profiled with DNA seqFISH+. c, d, Scaling of median spatial distance as a function of genomic distance for 20 chromosomes with 1-Mb resolution data in c and 25-kb resolution data in d. Grey dots represent the median distance of the given pairs of loci. Blue dashed lines are the median spatial distance at each genomic distance bin, while red lines are power-law function fits with the fitting parameters in the plots. n = 446 cells. e, The full spatial proximity map between all loci from the 1-Mb DNA seqFISH+ data with a search radius of 1 μm (bottom left triangle panel). The zoomed in view of the map for chr6 and chr7 (top right panel), showing the non-repetitive regions near pericentromeric repetitive regions from different chromosomes are more likely to be spatially close to each other. Colour bar is shown in log scale. f, Mean spatial proximity map for 20 chromosomes, considering only the first 5 Mb non-repetitive regions in each chromosome with a search radius of 1 μm. g, Distribution of coefficient of variation (CV) for spatial proximity from inter-chromosomal pairs in f. h, Single cell version of spatial proximity maps in f show heterogeneity in the spatial proximity between the proximal 5 Mb non-repetitive regions of the chromosomes. i, Single nuclei image shows that proximal 5 Mb non-repetitive regions from only a subset of chromosomes appear near the DAPI-rich pericentromeric heterochromatin regions in individual nuclei. The images are representative of n = 446 cells and the analysis are quantified from 2 biological replicates in e–h.
Extended Data Fig. 5 Visualization and validation for sequential immunofluorescence and repetitive element DNA FISH.
a, 17 antibodies and 4 repetitive elements, including gene-poor long interspersed nuclear elements (LINE1), gene-rich short interspersed nuclear elements (SINEB1), centromeric MinSat, and telomeres, are imaged along with DAPI. Individual cells have different patterns of immunofluorescence staining. Note that the DAPI patterns are not identical between cells. Similarly, marks that are colocalized with DAPI-rich pericentromeric heterochromatin regions are different between cells and even between different pericentromeric regions in a single cell. b, Representative H3K9ac image and edge-transformed image that detects the voxels on the exterior of H3K9ac globules (Methods). c, Representative H3K9ac images from a single z-section or maximum intensity z-projection with the intensity Z-score threshold above 2. 3D visualization (right) was performed for the pixels with the intensity Z-score above 2 (Methods). d, Additional single cell 3D images of immunofluorescence markers for the pixels with the intensity Z-score above 2. Heterochromatin components (H3K9me3, DAPI and MinSat) were clustered together, while RNAPIISer5-P, active marks (H3K9ac, H3K27ac), SINEB1 and nuclear speckles (SF3A66) were physically proximal. High intensity pixels of LINE1 by DNA FISH localized mainly to the LINE1-rich X chromosome88. e, Correlation of chromatin profiles for all 2,460 loci at 1 Mb resolution generated from distance to the interior and exterior voxels of different immunofluorescence marks (n = 446 cells). f, Scatter plots of the distances from each locus to interior voxels versus exterior voxels that are 2× s.d. above the mean for 2,460 loci at 1-Mb resolution (n = 446 cells). PCCs are shown. g, Heat map showing fraction of loci within 300 nm from immunofluorescence marks and repetitive elements by DNA seqFISH+ at 25-kb resolution (n = 1,200 loci and 446 cells).
Extended Data Fig. 6 Additional visualization and validation for fixed loci and chromatin profiles.
a, Correlation matrix comparing the chromatin profiles by DNA seqFISH+ and immunofluorescence with other methods7,24,35. The 1-Mb DNA seqFISH+ data were used and the reference data were binned with 1-Mb data. Chromatin profiles were computed as the fraction of loci within 300 nm from immunofluorescence marker exterior for the 2,460 loci (n = 446 cells). b, Two-dimensional density plots of individual marker comparison shown in a. n = 2,460 loci. c, Comparison of fraction of loci within 300 nm from the lamin B1 exterior with different thresholding values (Z-score above 2 or 3), or from nuclear periphery computed from convex hull of nuclear pixels (Methods), showing the good agreement of the profiles in different quantification criteria (n = 2,460 loci from 446 cells). d, Validation of lamin B1 enrichment with loci categorized as cell-type invariant constitutive lamina-associated domains (cLADs), cell-type dependent facultative LADs (fLADs), and constitutive inter-LADs (ciLADs) assigned from previous DamID studies35,89. Loci categorized as both cLADs and fLADs show enrichment of proximities to lamin B1 compared to those from ciLADs, representing a good agreement of our measurement (n = 351, 405 and 1,023 loci in cLADs, fLADs, ciLADs, respectively, category averaged from 446 cells) with the DamID studies. n is the number of loci. For the box plots in d and g, the centre line in the boxes marks median, the upper and lower limits of the boxes mark the interquartile range, the whiskers extend to the farthest data points within 1.5 times the interquartile range, and the grey points mark outliers. e, Additional visualization for chromatin profiles of Lamin B1 with different criteria in c (n = 446 cells) in comparison with Lamin B1 DamID profile35. To take into account only Lamin B1 staining at the nuclear periphery, we calculated the distances between the DNA loci and the Lamin B1 signal near the convex hull of the nucleus as well as with different intensity thresholds. f, Additional examples for single cell chromatin profiles in comparison with ChIP–seq24 for H3K27me3 (top) and SPRITE7. The profiles were computed and are displayed in the same way as Fig. 2c. n = 446 cells. g, The fraction of loci in single cells that are associated with exteriors of immunofluorescence markers for the fixed loci defined based on the chromatin profiles (n = 446 cells). Note that different immunofluorescence markers have different thresholds for calling fixed loci. Thus, fixed loci for some immunofluorescence markers are more consistently associated with the immunofluorescence marks in single cells. h, Additional 3D images of immunofluorescence markers and their associated fixed loci. In each cell, 6 immunofluorescence marks (2 per panel) are shown for visual clarity. i, 5 chromosomes are highlighted in the 3 cells shown in h. The fixed loci for a pair of immunofluorescence markers are shown for each chromosome in the corresponding image visualization. Fixed loci are shown in colored dots and the remaining loci on the chromosomes are shown as grey dots. The same colour codes are used in h.
Extended Data Fig. 7 Comparison between population level and single cell level chromosome organization in association with chromatin markers.
a, Clustering of the ensemble-averaged immunofluorescence spatial proximity profile of individual loci. n = 2,460 loci (n = 805, 278, 877, 500 loci in each cluster, respectively). b, In individual cells, loci associated with each cluster are mapped onto their spatial location. Note that cluster definitions for DNA loci were obtained from population-averaged data, and those cluster-assigned loci distribution may not necessarily reflect immunofluorescence marker localization in single cells. c, Box plot of immunofluorescence marks for the loci in each of the clusters. Cluster 1 is enriched in repressive markers such as H3K9me3, mH2A1, DAPI. Cluster 2 is enriched in interactions with fibrillarin. Cluster 3 is enriched in active marks such as RNAPII(Ser5P), H3K27ac and SF3A66 (nuclear speckle marker). Cluster 4 is enriched in lamin B1. For the box plots in c, d, h, i, the centre line in the boxes marks median, the upper and lower limits of the boxes mark the interquartile range, the whiskers extend to the farthest data points within 1.5 times the interquartile range, and the grey points mark outliers. d, The probability of loci of certain cluster pairs within 1 μm search radius in individual cells. Cluster definitions follow those in a–c. Randomized data were generated by scrambling the cluster identities of individual loci in cells while keeping the total number of loci within each cluster the same within that cell. The probability for observed and randomized data for each cell are shown as box plots. e, The probability that pairs of loci with cluster assignments are found within a given search radius, as a function of search radius. Error bars represent standard error over 20 bootstrap trials. f, Mapping of the A/B compartment definitions23 onto the t-SNE plot based on the ensemble-averaged loci-immunofluorescence mark spatial proximity map. Note that regions that are not assigned to one of the compartments were excluded from the analysis. (n = 1,188 and 960 loci in A and B compartments, respectively). g, Reconstructions of individual cells with loci assigned as A or B compartment mapped onto their spatial location. Observed compared to randomized data for 2 cells shown in b. h, Box plot of the immunofluorescence marks for the loci assigned to A or B compartments. i, The probability that loci in A/B compartments are within 1 μm search radius in individual cells, similar to d. j, The probability that pairs of loci with A/B assignments are found within a given search radius, as a function of search radius for spatial proximity, similar to e. n = 446 cells from two biological replicates in a–j.
Extended Data Fig. 8 Further characterization of nuclear zones and interfaces.
a, Analysis workflow for the pixel-based combinatorial chromatin profiling. Individual voxels with the 15 chromatin markers are clustered with hierarchical clustering and visually represented by a nonlinear dimensionality reduction technique (UMAP)70. Voxels from individual clusters (zones) are mapped back to individual nuclei, and overlaid with DNA seqFISH+ dots. b, UMAP representation for 44,000 pixels sampled from 201 cells, labelled with 12 zones. UMAP projection is used for visual clarity. c, Pearson correlation matrix between zones and interfaces based on the DNA loci association with zones and interfaces shown in f (n = 2,460 loci). Loci appearing in zone 1 are also more likely to be found in zone 2 as well as in interface 1/2. d, Comparison of zone appearance with and without DNA seqFISH+ treatment shows an overall agreement between the measurements. Mean values from 20 bootstrap trials are shown with error bars corresponding to standard errors. e, Assignment of zones as a function of downsampling of immunofluorescence markers. Twenty random subsets of immunofluorescence markers are selected at each downsample size. The centre of the curve reflects the mean and the width reflects the standard deviation of the correct zone assignments at each downsample size (Methods). f, Reconstructions of zones and DNA loci in additional cells. g, Reconstructions of zones in the cell 31 with different z-planes. h, Reconstruction of zones and 1,000-gene intron dots as well as RNAPIISer5-P staining (background-subtracted) and edge of RNAPIISer5-P staining. i, Heat map for probability of association between DNA loci, nuclear zones and interfaces for the 1-Mb data. Zones and interfaces are ordered according to the overall probability of association with DNA loci. Right panel shows the loci around Pou5f1 visualized in Fig. 3b (panel 1). Each locus in single cells is assigned to one zone or interface. The distribution shown in the heat map reflects the single cell variability in zone association for each locus. For example, Ehmt2 and Pou5f1 loci were primarily associated with active zone 2 and interfaces 1/2 and 2/3, whereas Opn5 and Dazl loci were more uniformly distributed across many zones. j, Heat map for probability of association between DNA loci, nuclear zones and interfaces for the 25-kb data. Loci within the same Mb region have similar nuclear zone and interface association probability. k, Frequency of association between DNA loci and zones or interfaces in single cells, calculated for all loci, loci with intra-chromosomal and interchromosomal pairs, TAS measured by intron FISH, and random loci (randomized control). Mean values from 20 bootstrap trials are shown with error bars corresponding to standard errors. l, Correlation between zone association and gene expression levels (RNA-seq)49, density of RNA polymerases on the loci (GRO-seq)75 and early replication domains (Repli-seq)76 for all loci at 1-Mb resolution (n = 2,460 loci). m, Expression levels of fixed loci for each immunofluorescence marker from n = 446 cells. Population level expressions are taken from bulk RNA-seq studies49 and integrated for a 1-Mb region. For the box plots, the centre line in the boxes marks median, the upper and lower limits of the boxes mark the interquartile range, the whiskers extend to the farthest data points within 1.5 times the interquartile range, and the grey points mark outliers. n, Correlation of mRNA levels and fraction of voxels within 300 nm of a given locus in single cells being in active zones for individual mRNAs. Mean values from 20 bootstrap trials are shown with error bars corresponding to standard errors for each mRNA. Randomized samples correspond to scrambling of mRNA and zone assignment values for each cell. o, Comparison of fraction of voxels within 300 nm of DNA loci to be in active zones (zone 1 and 2) for loci with an active intron signal (ON) versus loci with no intron signal (OFF) for individual introns. Mean values from 20 bootstrap trials are shown with error bars corresponding to standard errors. for each intronic RNA. n = 201 and 172 cells for DNA seqFISH+ and intron FISH measurements in b–l, n, o, respectively.
Extended Data Fig. 9 Heterogeneity of transcriptional and chromatin states and their relationships in single cells.
a, Pearson correlation of mean mRNA counts by RNA seqFISH and bulk RNA-seq. Error bars for RNA seqFISH represent the standard error of the mean from two measurements (n = 151 and 175 cells from the centre field of views). b, UMAP representation of individual cells in two different cell clusters identified based on scRNA-seq42 and mapped onto RNA seqFISH data (cluster a for cells with more pluripotent states and cluster b for cells on the differentiation path) (left), and in different datasets (right) (n = 326 and 250 cells for RNA seqFISH and scRNA-seq42 dataset, respectively). c, Box plots showing a good agreement of differentially expressed genes in scRNA-seq and seqFISH datasets. P values were from a two-sided Wilcoxon’s rank-sum test with cells in clusters a and b (n = 298 and 209 cells in cluster a and n = 28 and 41 cells in cluster b with RNA seqFISH and scRNA-seq42 datasets, respectively). For the box plots, the centre line in the boxes marks median, the upper and lower limits of the boxes mark the interquartile range, the whiskers extend to the farthest data points within 1.5 times the interquartile range, and the grey points mark outliers. d, UMAP representations of the cell clusters defined by immunofluorescence intensity profiles. e, Heat map of cell clusters with distinct immunofluorescence profiles shown with cell cycle associated immunofluorescence markers and all mRNA markers, similar to Fig. 4b. f, Pseudotime-course analysis for cell cycle progression, cell cycle markers (H4K16ac, H4K20me1 and H3pSer10) show clear enrichments while other markers do not show specific enrichments upon cell cycle pseudotime course, suggesting majority of the immunofluorescence markers profiled are not primarily affected by cell cycle phases. g, Pseudotimecourse analysis for pluripotency states in mouse ES cells based on scaled mRNA expression levels, showing the enrichment from markers associated with naive pluripotency such as Tfcp2l1 and Nanog to markers associated with primed pluripotency such as Dnmt3a, Lin28b and Otx2 as well as the enrichment of certain chromatin marks upon the pluripotency pseudotime course. h, Scaled marker gene expression (top panels) or intensity (bottom panels) along the pluripotency pseudotime ordering of cells. Raw data in g are overlaid with fitting curves (Methods). i, Network analysis for the mRNA and immunofluorescence markers represents positive and negative Pearson correlation relationships among markers. j, Joint Pearson correlation matrix between mRNA and immunofluorescence markers based on the scaled expression or intensity profiles in single cells (n = 41 mRNA and 25 immunofluorescence markers). n = 326 cells in the centre field of views for RNA seqFISH and immunofluorescence data in a–j.
Extended Data Fig. 10 Additional analysis for colony level cell state heterogeneity.
a, mRNA and immunofluorescence images in a colony in the 48-h clonal tracing experiment. H3K27me3 and mH2A1 overall intensities are similar in WT cells (GFP/Neo negative) in the colony. b, Standard deviation of normalized mRNA levels within colonies (red) and between colonies (grey). Error bars show standard errors for 20 bootstrap trials. Tbx3 and Nanog are more homogeneous within colonies, consistent with previous findings of the long-lived transcriptional states of these genes across several generations by single-cell live-imaging experiments41,49. n = 117 unlabelled cells within colonies from a 48-h dataset. c, Histogram of cell-to-cell correlations of chromosome to chromosome proximity maps for cells within colonies (red) and between colonies (grey). Cells with similar chromosome structures (red dots with high correlation values) are likely to be sister cells. The y-axis represents PCC, computed by 20 × 20 chromosome proximity matrices from pairs of cells. P values were from a two-sided Wilcoxon’s rank-sum test with pairs of cells of 180, 1,198, 966 and 5,820 (left to right). d, Correlation of chromosome proximities between cells in colonies in the 48-h clonal tracing experiment. Strong correlations are seen between putative sister cells suggesting that gross chromosome proximities are preserved for one generation. Colour bars represent PCC computed in c. e, Chromosome images for unlabelled cells from a 24-h colony shows similarities between two sets of neighbouring cells (maximum z-projection). Chromosome organizations in single cells are highly correlated between pairs of cells that were physically close (possibly sister cells) and are mostly uncorrelated with other cells in the colonies. Six chromosomes are shown for visual clarity. r represents PCC computed in c.
Supplementary information
Supplementary Table
Supplementary Table 1: List for genomic coordinates of the 3,660 DNA loci used in DNA seqFISH+.
Supplementary Table
Supplementary Table 2: Codebook for the 3,660 DNA loci in the three fluorescent channels. Base 16 pseudocolor coding scheme for each of the loci in the channel 1 and 2, include control regions for off target evaluation. Region and chromosome paint imaging scheme for each of the loci in the channel 3.
Supplementary Table
Supplementary Table 3: Chromosome configuration probability of finding fixed points for a pair of IF markers on a given chromosome simultaneously in single cells. Fraction of loci associated within 300 nm of interior and exterior voxels of each IF marker in single cells. Median spatial distance from each of the 3,660 DNA loci to interior and exterior voxels of individual immunofluorescence markers.
Supplementary Table
Supplementary Table 4: Normalized association frequencies of individual zones and interfaces on each of the 3,660 DNA loci.
Supplementary Table
Supplementary Table 5: List for fraction of DNA loci associated with zones/interfaces in single cells, for all loci, intra-chromosomal and inter-chromosomal pairs of loci, transcriptional active sites measured by intron FISH, and randomly positioned spots. p-values for pairs of categories were computed from 20 bootstrap trials with a two-sided Wilcoxon’s rank sum test.
Supplementary Table
Supplementary Table 6: List for target RNAs in E14 replicates and clonal experiments.
Rights and permissions
About this article

Cite this article
Takei, Y., Yun, J., Zheng, S. et al. Integrated spatial genomics reveals global architecture of single nuclei. Nature 590, 344–350 (2021). https://doi.org/10.1038/s41586-020-03126-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-03126-2
This article is cited by
-
pyHiM: a new open-source, multi-platform software package for spatial genomics based on multiplexed DNA-FISH imaging
Genome Biology (2024)
-
Spatial multi-omics: novel tools to study the complexity of cardiovascular diseases
Genome Medicine (2024)
-
Computational methods for analysing multiscale 3D genome organization
Nature Reviews Genetics (2024)
-
Polycomb repression of Hox genes involves spatial feedback but not domain compaction or phase transition
Nature Genetics (2024)
-
Simultaneous single-cell three-dimensional genome and gene expression profiling uncovers dynamic enhancer connectivity underlying olfactory receptor choice
Nature Methods (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
