
Abstract
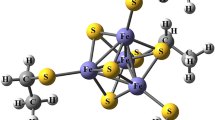
The fine structures of proteins, such as the positions of hydrogen atoms, distributions of valence electrons and orientations of bound waters, are critical factors for determining the dynamic and chemical properties of proteins. Such information cannot be obtained by conventional protein X-ray analyses at 3.0–1.5 Å resolution, in which amino acids are fitted into atomically unresolved electron-density maps and refinement calculations are performed under strong restraints1,2. Therefore, we usually supplement the information on hydrogen atoms and valence electrons in proteins with pre-existing common knowledge obtained by chemistry in small molecules. However, even now, computational calculation of such information with quantum chemistry also tends to be difficult, especially for polynuclear metalloproteins3. Here we report a charge-density analysis of the high-potential iron–sulfur protein from the thermophilic purple bacterium Thermochromatium tepidum using X-ray data at an ultra-high resolution of 0.48 Å. Residual electron densities in the conventional refinement are assigned as valence electrons in the multipolar refinement. Iron 3d and sulfur 3p electron densities of the Fe4S4 cluster are visualized around the atoms. Such information provides the most detailed view of the valence electrons of the metal complex in the protein. The asymmetry of the iron–sulfur cluster and the protein environment suggests the structural basis of charge storing on electron transfer. Our charge-density analysis reveals many fine features around the metal complex for the first time, and will enable further theoretical and experimental studies of metalloproteins.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


References
Hendrickson, W. A. Stereochemically restrained refinement of macromolecular structures. Methods Enzymol. 115, 252–270 (1985)
Wlodawer, A., Minor, W., Dauter, Z. & Jaskolski, M. Protein crystallography for aspiring crystallographers or how to avoid pitfalls and traps in macromolecular structure determination. FEBS J. 280, 5705–5736 (2013)
Rokob, T. A., Srnec, M. & Rulíšek, L. Theoretical calculations of physico-chemical and spectroscopic properties of bioinorganic systems: current limits and perspectives. Dalton Trans. 41, 5754–5768 (2012)
Nogi, T., Fathir, I., Kobayashi, M., Nozawa, T. & Miki, K. Crystal structures of photosynthetic reaction center and high-potential iron-sulfur protein from Thermochromatium tepidum: thermostability and electron transfer. Proc. Natl Acad. Sci. USA 97, 13561–13566 (2000)
Liu, L., Nogi, T., Kobayashi, M., Nozawa, T. & Miki, K. Ultrahigh-resolution structure of high-potential iron-sulfur protein from Thermochromatium tepidum. Acta Crystallogr. D 58, 1085–1091 (2002)
Niwa, S. et al. Structure of the LH1-RC complex from Thermochromatium tepidum at 3.0 Å. Nature 508, 228–232 (2014)
Takeda, K., Kusumoto, K., Hirano, Y. & Miki, K. Detailed assessment of X-ray induced structural perturbation in a crystalline state protein. J. Struct. Biol. 169, 135–144 (2010)
Niu, S. & Ichiye, T. Insight into environmental effects on bonding and redox properties of [4Fe-4S] clusters in proteins. J. Am. Chem. Soc. 131, 5724–5725 (2009)
Glaser, T. et al. Protein effects on the electronic structure of the [Fe4S4]2+ cluster in ferredoxin and HiPIP. J. Am. Chem. Soc. 123, 4859–4860 (2001)
Dey, A. et al. Solvent tuning of electrochemical potentials in the active sites of HiPIP versus ferredoxin. Science 318, 1464–1468 (2007)
Jelsch, C. et al. Accurate protein crystallography at ultra-high resolution: valence electron distribution in crambin. Proc. Natl Acad. Sci. USA 97, 3171–3176 (2000)
Schmidt, A., Jelsch, C., Ostergaard, P., Rypniewski, W. & Lamzin, V. S. Trypsin revisited: crystallography at (sub) atomic resolution and quantum chemistry revealing details of catalysis. J. Biol. Chem. 278, 43357–43362 (2003)
Fournier, B. et al. Charge density and electrostatic interactions of fidarestat, an inhibitor of human aldose reductase. J. Am. Chem. Soc. 131, 10929–10941 (2009)
Zarychta, B. et al. Cholesterol oxidase: ultrahigh-resolution crystal structure and multipolar atom model-based analysis. Acta Crystallogr. D 71, 954–968 (2015)
Hansen, N. K. & Coppens, P. Testing aspherical atom refinements on small-molecule data sets. Acta Crystallogr. A 34, 909–921 (1978)
Berkholz, D. S., Driggers, C. M., Shapovalov, M. V., Dunbrack, R. L., Jr & Karplus, P. A. Nonplanar peptide bonds in proteins are common and conserved but not biased toward active sites. Proc. Natl Acad. Sci. USA 109, 449–453 (2012)
Engh, R. A. & Huber, R. Accurate bond and angle parameters for X-ray structure refinement. Acta Crystallogr. A 47, 392–400 (1991)
Murray-Rust, P. & Glusker, J. P. Directional hydrogen bonding to sp2- and sp3- hybridized oxygen atoms and its relevance to ligand-macromolecule interactions. J. Am. Chem. Soc. 106, 1018–1025 (1984)
Bertini, I., Donaire, A., Felli, I. C., Luchinat, C. & Rosato, A. 1H and 13C NMR studies of an oxidized HiPIP. Inorg. Chem. 36, 4798–4803 (1997)
Improta, R., Vitagliano, L. & Esposito, L. Peptide bond distortions from planarity: new insights from quantum mechanical calculations and peptide/protein crystal structures. PLoS ONE 6, e24533 (2011)
Wang, Y.-F., Yu, Z.-Y., Wu, J. & Liu, C.-B. Electron delocalization and charge transfer in polypeptide chains. J. Phys. Chem. A 113, 10521–10526 (2009)
Smith, G. T. et al. Experimental determination of the electron density topology in a non-centrosymmetric transition metal complex: [Ni(H3L)][NO3][PF6] [H3L = N,N′,N′′-tris(2-hydroxy-3-methylbutyl)-1,4,7-triazacyclononane]. J. Am. Chem. Soc. 119, 5028–5034 (1997)
Bader, R. F. W. Atoms in Molecules: A Quantum Theory (Oxford Univ. Press, 1990)
Koritsanszky, T. S. & Coppens, P. Chemical applications of X-ray charge-density analysis. Chem. Rev. 101, 1583–1628 (2001)
Gibbs, G. V. et al. Theoretical electron density distributions for Fe- and Cu-sulfide earth materials: a connection between bond length, bond critical point properties, local energy densities, and bonded interactions. J. Phys. Chem. B 111, 1923–1931 (2007)
Harris, T. V. & Szilagyi, R. K. Iron-sulfur bond covalency from electronic structure calculations for classical iron-sulfur clusters. J. Comput. Chem. 35, 540–552 (2014)
Dey, A. et al. Sulfur K-edge XAS and DFT calculations on [Fe4S4]2+ clusters: effects of H-bonding and structural distortion on covalency and spin topology. Inorg. Chem. 44, 8349–8354 (2005)
Mouesca, J. M., Noodleman, L., Case, D. A. & Lamotte, B. Spin densities and spin coupling in iron-sulfur clusters: a new analysis of hyperfine coupling constants. Inorg. Chem. 34, 4347–4359 (1995)
Flot, D. et al. The ID23-2 structural biology microfocus beamline at the ESRF. J. Synchrotron Radiat. 17, 107–118 (2010)
Paithankar, K. S., Owen, R. L. & Garman, E. F. Absorbed dose calculations for macromolecular crystals: improvements to RADDOSE. J. Synchrotron Radiat. 16, 152–162 (2009)
Otwinowski, Z. & Minor, W. Processing of X-ray diffraction data. Methods Enzymol. 276, 307–326 (1997)
Sheldrick, G. M. A short history of SHELX. Acta Crystallogr. A 64, 112–122 (2008)
Guillot, B., Viry, L., Guillot, R. & Lecomte, C. Refinement of proteins at subatomic resolution with MOPRO. J. Appl. Cryst. 34, 214–223 (2001)
Guillot, B., Jelsch, C., Podjarny, A. & Lecomte, C. Charge-density analysis of a protein structure at subatomic resolution: the human aldose reductase case. Acta Crystallogr. D 64, 567–588 (2008)
Allen, F. H. A systematic pairwise comparison of geometric parameters obtained by X-ray and neutron diffraction. Acta Crystallogr. B 42, 515–522 (1986)
Wu, G., Rodrigues, B. L. & Coppens, P. The correction of reflection intensities for incomplete absorption of high-energy X-rays in the CCD phosphor. J. Appl. Cryst. 35, 356–359 (2002)
Zarychta, B., Pichon-Pesme, V., Guillot, B., Lecomte, C. & Jelsch, C. On the application of an experimental multipolar pseudo-atom library for accurate refinement of small-molecule and protein crystal structures. Acta Crystallogr. A 63, 108–125 (2007)
Holladay, A., Leung, P. & Coppens, P. Generalized relations between d-orbital occupancies of transition-metal atoms and electron-density multipole population parameters from X-ray diffraction data. Acta Crystallogr. A 39, 377–387 (1983)
Sabino, J. R. & Coppens, P. On the choice of d-orbital coordinate system in charge-density studies of low-symmetry transition-metal complexes. Acta Crystallogr. A 59, 127–131 (2003)
DeLano, W. L. The PyMol Molecular Graphics System (DeLano Scientific, 2002)
Yu, M. & Trinkle, D. R. Accurate and efficient algorithm for Bader charge integration. J. Chem. Phys. 134, 064111 (2011)
Acknowledgements
We thank K. Kusumoto and H. Ohno for their contributions in the initial steps of the work, and T. Tsujinaka and S. Niwa for their contributions in the preparation of the manuscript. We also thank the BL41XU beamline staff of SPring-8 for their help in data collection. This work was supported by a Grant-in-Aid for Scientific Research (number 23657073 to K.T.) and the Photon and Quantum Basic Research Coordinated Development Program (to K.M.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Author information
Authors and Affiliations
Contributions
K.M. initiated and supervised the project. K.T. designed the experiments. Y.H. prepared crystals. Y.H. and K.T. performed data collection and the crystallographic analysis. Y.H., K.T. and K.M. discussed the results. Y.H. wrote the initial draft, and K.T. and K.M. revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 Quality of the diffraction data at 0.48 Å resolution.
a, The diffraction image. Right, zoom view of the boxed region at left. The resolution for each circle is indicated. b, Rsym (blue) and <I>/<σ(I)> (pink) values are plotted for 30 resolution bins. c, Changes of Rsym at the highest-resolution shell (0.50–0.48 Å) and relative B factor in the course of the data collection. The Rsym (blue) and relative B factor (red) are plotted as functions of frame number.
Extended Data Figure 2 Residual electron density for each refinement step.
The left panels show the residual density after the ISAM refinement; the right panels show the residual density after the MAM refinement. a, The plane of the peptide bond between Asn45 and Cys46. Maximum and minimum peaks are 0.33 and −0.22 electrons per cubic ångström for the ISAM analysis, and 0.18 and −0.20 electrons per cubic ångström for the MAM analysis. b, The plane of the aromatic ring of Trp74. Maximum and minimum peaks are 0.34 and −0.29 electrons per cubic ångström for the ISAM analysis, and 0.23 and −0.23 electrons per cubic ångström for the MAM analysis. c, The Fe4S4 cluster. The plane consists of FE1, S3 and Cys43-Sγ atoms. Maximum and minimum peaks are 0.60 and −0.35 electrons per cubic ångström for the ISAM analysis, and 0.35 and −0.29 electrons per cubic ångström for the MAM analysis. The contour interval is 0.05 electrons per cubic ångström for all figures. Blue solid, red dashed and yellow dashed lines denote positive, negative and zero contours, respectively.
Extended Data Figure 3 Interaction network around the Fe4S4 cluster.
a, Deformation electron density around the Cys43-Sγ atom. The main-chain oxygen atom of Asn70, the main-chain carboxyl carbon atom of Gly73 and the H atom of Ile69-Cδ1 are located close to Cys43-Sγ. The static deformation maps are shown as grey and cyan surfaces contoured at the levels of +0.1 and +0.3 electrons per cubic ångström, respectively. The omit map of hydrogen atoms is shown as a pink mesh contoured at the 3.0σ level. The dashed lines indicate interactions between valence densities of sulfur atoms and hydrogen atoms. b, Deformation electron density around Cys61-Sγ. The main-chain amide of Leu63 and the H atom of Phe64-Cδ2 are located close to Cys61-Sγ. c, Deformation electron density around Cys75-Sγ. The main-chain amide of Ser77 is located close to Cys75-Sγ. d, Deformation electron density around S1 of the Fe4S4 cluster. The H atom of Phe48-Cδ2 and the Cδ1 atom of Leu63 are located close to S1. e, Deformation electron density around S2 of the Fe4S4 cluster. The H atoms of Tyr19-Cδ1, Phe64-Cε2 and Ile69-Cγ2 are located close to S2. f, Deformation electron density around S4 of the Fe4S4 cluster. The H atom of Cys43-Cβ, the H atom of Cys46-Cβ and the amide nitrogen atom of Met49 are located close to S4.
Extended Data Figure 4 The local axes for Fe atoms of Fe4S4(Cys-Sγ)4.
a, Whole view of the local axes of the four Fe atoms. b, Close-up views of the local axes of each Fe atom (FE1−FE4). The static deformation maps of Fe4S4(Cys-Sγ)4 are represented as grey isosurfaces contoured at the level of +0.2 electrons per cubic ångström.
Rights and permissions
About this article

Cite this article
Hirano, Y., Takeda, K. & Miki, K. Charge-density analysis of an iron–sulfur protein at an ultra-high resolution of 0.48 Å. Nature 534, 281–284 (2016). https://doi.org/10.1038/nature18001
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature18001
This article is cited by
-
Prediction of hydrophilic and hydrophobic hydration structure of protein by neural network optimized using experimental data
Scientific Reports (2023)
-
Measurement of charges and chemical bonding in a cryo-EM structure
Communications Chemistry (2023)
-
Structural and molecular properties of complexes of biomolecules and metal–organic frameworks: dispersion-corrected DFT treatment
Journal of Molecular Modeling (2022)
-
Crystal structure of a photosynthetic LH1-RC in complex with its electron donor HiPIP
Nature Communications (2021)
-
Regulatory mechanisms of ryanodine receptor/Ca2+ release channel revealed by recent advancements in structural studies
Journal of Muscle Research and Cell Motility (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
