
Abstract
Naive embryonic stem cells hold great promise for research and therapeutics as they have broad and robust developmental potential. While such cells are readily derived from mouse blastocysts it has not been possible to isolate human equivalents easily1,2, although human naive-like cells have been artificially generated (rather than extracted) by coercion of human primed embryonic stem cells by modifying culture conditions2,3,4 or through transgenic modification5. Here we show that a sub-population within cultures of human embryonic stem cells (hESCs) and induced pluripotent stem cells (hiPSCs) manifests key properties of naive state cells. These naive-like cells can be genetically tagged, and are associated with elevated transcription of HERVH, a primate-specific endogenous retrovirus. HERVH elements provide functional binding sites for a combination of naive pluripotency transcription factors, including LBP9, recently recognized as relevant to naivety in mice6. LBP9–HERVH drives hESC-specific alternative and chimaeric transcripts, including pluripotency-modulating long non-coding RNAs. Disruption of LBP9, HERVH and HERVH-derived transcripts compromises self-renewal. These observations define HERVH expression as a hallmark of naive-like hESCs, and establish novel primate-specific transcriptional circuitry regulating pluripotency.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Change history
17 December 2014
Cell line hiPS-SK4 was corrected to hFF-iPS4 in Fig. 1, Methods and the Acknowledgements.
References
Welling, M. & Geijsen, N. Uncovering the true identity of naive pluripotent stem cells. Trends Cell Biol. 23, 442–448 (2013)
Ware, C. B. et al. Derivation of naïve human embryonic stem cells. Proc. Natl Acad. Sci. 111, 4484–4489 (2014)
Chan, Y. S. et al. Induction of a human pluripotent state with distinct regulatory circuitry that resembles preimplantation epiblast. Cell Stem Cell 13, 663–675 (2013)
Gafni, O. et al. Derivation of novel human ground state naive pluripotent stem cells. Nature 504, 282–286 (2013)
Hanna, J. et al. Human embryonic stem cells with biological and epigenetic characteristics similar to those of mouse ESCs. Proc. Natl Acad. Sci. USA 107, 9222–9227 (2010)
Martello, G., Bertone, P. & Smith, A. Identification of the missing pluripotency mediator downstream of leukaemia inhibitory factor. EMBO J. 32, 2561–2574 (2013)
Kunarso, G. et al. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nature Genet. 42, 631–634 (2010)
Lu, X. et al. The retrovirus HERVH is a long noncoding RNA required for human embryonic stem cell identity. Nature Struct. Mol. Biol. 21, 423–425 (2014)
Fort, A. et al. Deep transcriptome profiling of mammalian stem cells supports a regulatory role for retrotransposons in pluripotency maintenance. Nature Genet. 46, 558–566 (2014)
Macfarlan, T. S. et al. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 487, 57–63 (2012)
Santoni, F. A., Guerra, J. & Luban, J. HERV-H RNA is abundant in human embryonic stem cells and a precise marker for pluripotency. Retrovirology 9, 111 (2012)
Kelley, D. & Rinn, J. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol. 13, R107 (2012)
Gaspar-Maia, A. et al. Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature 460, 863–868 (2009)
Chappell, J., Sun, Y., Singh, A. & Dalton, S. MYC/MAX control ERK signaling and pluripotency by regulation of dual-specificity phosphatases 2 and 7. Genes Dev. 27, 725–733 (2013)
Xie, W. et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 153, 1134–1148 (2013)
van den Berg, D. L. et al. An Oct4-centered protein interaction network in embryonic stem cells. Cell Stem Cell 6, 369–381 (2010)
Chen, X. et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133, 1106–1117 (2008)
Loewer, S. et al. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nature Genet. 42, 1113–1117 (2010)
Ng, S. Y., Johnson, R. & Stanton, L. W. Human long non-coding RNAs promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. EMBO J. 31, 522–533 (2012)
Ye, S., Li, P., Tong, C. & Ying, Q. L. Embryonic stem cell self-renewal pathways converge on the transcription factor Tfcp2l1. EMBO J. 32, 2548–2560 (2013)
Nishiyama, A. et al. Systematic repression of transcription factors reveals limited patterns of gene expression changes in ES cells. Sci. Rep. 3, 1390 (2013)
Mátés, L. et al. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nature Genet. 41, 753–761 (2009)
Ramsköld, D. et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nature Biotechnol. 30, 777–782 (2012)
Yan, L. Y. et al. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nature Struct. Mol. Biol. 20, 1131–1139 (2013)
Nichols, J. & Smith, A. Naive and primed pluripotent states. Cell Stem Cell 4, 487–492 (2009)
Okamoto, I. et al. Eutherian mammals use diverse strategies to initiate X-chromosome inactivation during development. Nature 472, 370–374 (2011)
Theunissen, T. W. et al. Systematic identification of culture conditions for induction and maintenance of naive human pluripotency. Cell Stem Cell http://dx.doi.org/10.1016/j.stem.2014.07.002 (2014)
To, S., Rodda, S. J., Rathjen, P. D. & Keough, R. A. Modulation of CP2 family transcriptional activity by CRTR-1 and sumoylation. PLoS ONE 5, e11702 (2010)
Dunn, S. J., Martello, G., Yordanov, B., Emmott, S. & Smith, A. G. Defining an essential transcription factor program for naive pluripotency. Science 344, 1156–1160 (2014)
Grabundzija, I. et al. Sleeping Beauty transposon-based system for cellular reprogramming and targeted gene insertion in induced pluripotent stem cells. Nucleic Acids Res. 41, 1829–1847 (2013)
Haase, A. et al. Generation of induced pluripotent stem cells from human cord blood. Cell Stem Cell 5, 434–441 (2009)
Prigione, A., Fauler, B., Lurz, R., Lehrach, H. & Adjaye, J. The senescence-related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells 28, 721–733 (2010)
Takahashi, K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007)
Onder, T. T. et al. Chromatin-modifying enzymes as modulators of reprogramming. Nature 483, 598–602 (2012)
Ivics, Z., Hackett, P. B., Plasterk, R. H. & Izsvak, Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 91, 501–510 (1997)
Kaufman, C. D., Izsvak, Z., Katzer, A. & Ivics, Z. Frog Prince transposon-based RNAi vectors mediate efficient gene knockdown in human cells. J. RNAi Gene Silencing 1, 97–104 (2005)
Wang, Z., Oron, E., Nelson, B., Razis, S. & Ivanova, N. Distinct lineage specification roles for NANOG, OCT4, and SOX2 in human embryonic stem cells. Cell Stem Cell 10, 440–454 (2012)
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013)
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009)
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013)
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods 9, 357–359 (2012)
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008)
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014)
Ernst, J. & Kellis, M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nature Biotechnol. 28, 817–825 (2010)
Pohl, A. & Beato, M. bwtool: a tool for bigWig files. Bioinformatics 30, 1618–1619 (2014)
Frith, M. C. et al. Detection of functional DNA motifs via statistical over-representation. Nucleic Acids Res. 32, 1372–1381 (2004)
Haverty, P. M., Hansen, U. & Weng, Z. Computational inference of transcriptional regulatory networks from expression profiling and transcription factor binding site identification. Nucleic Acids Res. 32, 179–188 (2004)
Neph, S. et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 489, 83–90 (2012)
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010)
Volders, P.-J. et al. LNCipedia: a database for annotated human lncRNA transcript sequences and structures. Nucleic Acids Res. 41, 246–251 (2012)
Kong, L. et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 35, W345–W349 (2007)
Vassena, R. et al. Waves of early transcriptional activation and pluripotency program initiation during human preimplantation development. Development 138, 3699–3709 (2011)
Hanna, J. et al. Metastable pluripotent states in NOD-mouse-derived ESCs. Cell Stem Cell 4, 513–524 (2009)
Bellucci, M., Agostini, F., Masin, M. & Tartaglia, G. G. Predicting protein associations with long noncoding RNAs. Nature Methods 8, 444–445 (2011)
Zhou, W. et al. Induction of human fetal globin gene expression by a novel erythroid factor, NF-E4. Mol. Cell. Biol. 20, 7662–7672 (2000)
Havugimana, P. C. et al. A census of human soluble protein complexes. Cell 150, 1068–1081 (2012)
Acknowledgements
L.D.H. is Wolfson Royal Society Research Merit Award Holder. A.T.G. is funded by a scholarship from the University of Bath. Z.Iz. is funded by ERC-2011-AdG 294742. G.G.S. is funded by DFG grant SCHU1014/8-1 and LOEWE Center for Cell and Gene Therapy Frankfurt/Hessian Ministry of Higher Education, Research and the Arts (ref. number III L 4-518/17.004). We thank U. Martin and S. Merkert (Leibniz Research Laboratories for Biotechnology and Artificial Organs (LEBAO), Hannover Medical School, Hannover, Germany) for providing the cell lines hCBEC, hCBiPS1, hCBiPS2 and hFF-iPS4. We thank G. Klein for the inspiration of working with ERVs and Z. Cseresnyés for his assistance in imaging.
Author information
Authors and Affiliations
Contributions
This project was inspired by M.C.L. Z.Iz., L.D.H. and J.W. conceived ideas for the project, and wrote the manuscript with contributions from other authors. The project was supervised by Z.Iz. and L.D.H. Z.Iv. provided critical advice. J.W. designed and performed experiments, analysed and interpreted data, and participated in bioinformatic analyses. T.R. contributed by EMSA and assisted in immunostaining experiments. A.S. assisted in the reporter assays. H.C. assisted in shRNA cloning. W.C. and J.W. performed RNA-seq experiments. A.P. provided materials and performed karyotype analysis. D.B., N.V.F. and G.G.S. provided materials. G.X. performed RNA-seq, bisulfite-seq and ChIP-seq analyses. M.S. analysed microarray data and performed cross-species correlation studies. L.D.H. and A.T.G. performed all the other bioinformatic analyses.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 HERVH is the most transcriptionally enriched transposable element in hPSCs.
a, Heat map showing expression of repetitive element classes in human induced pluripotent stem cells (hiPSCs), fibroblasts (HFF-1) and hiPSC-derived embryoid bodies (EBs). b, Highly expressed (top 20) LTR elements in hESCs (top panel) and hiPSCs (bottom panel). Red bars: proportion of reads of each LTR element in total LTR-element-related reads. Blue bars: enrichment of each LTR element relative to the background (calculation details described in Methods). c, d, Heat maps showing the expression profile of 1,225 full-length HERVHs in various human cell types. For a list of samples and expression data see Supplementary Tables 4 and 7, respectively. c, Expression profile of HERVH in 43 normal somatic tissues, 8 cancer cell lines and 55 hESC (H1, H6 and H9) and 26 hiPSC samples, including our hiPSC30 line. The rows represent the transcription from 1,225 full-length HERVH loci. d, Expression profile of HERVHs in hPSC lines and single cells from three individual hESC clones. On the basis of their expression, the 1,225 full-length HERVH loci are clustered into three groups (highly, moderately and inactive). Note that HERVH activity is heterogeneous between single cells of an hPSC population. e, HERVH expression in single hESCs positively correlates with the expression of key pluripotency-associated transcription factors. Note that SOX2, not illustrated, shows no correlation (P = 0.59). Each dot represents a single hESC sample24.
Extended Data Figure 2 HERVH shows the hallmarks of active chromatin in hPSCs.
a, Chromatin status analysis around full-length HERVHs in hESC_H1. The promoter/transcription initiation regions and the transcribed regions of active HERVH loci are associated with active epigenetic marks and chromatin modifiers. The neighbouring regions of inactive HERVH loci show the hallmarks of heterochromatin. b, Active HERVHs were enriched with binding sites for CHD1 compared to inactive ones. Chi-squared tests were performed, P values shown as statistical significance. c, Comparison of epigenetic marks and chromatin modifiers in proximity of HERVH internal sequence (HERVH-int) and LTR7. As a control, we employ HERVK-int and LTR5. We compare the number of marks within or near active and inactive versions (allowing 1.5 kb either side) of each element in ES cells. Expected numbers are derived from a null of no relative enrichment and P values determined by Chi-squared. *P < 0.05, **P < 0.01, ***P < 0.001 (for data see Supplementary Table 15). d, Cross-tissue comparison of the distance of the closest DHS to the active sequences not including any DHS. The distances are presented in log ratio. e, Pie charts show chromatin state segmentation for hESC_H1 in full-length HERVK/HML2 and HERVH regions. Most HERVK regions are repressed while a sub-population of HERVH loci is active. Chromatin status analysis of HERVK/HML2 loci reveals that transcription of the few activated HERVK loci is promoted primarily by neighbouring regulatory elements, and not by their own LTRs. The chromatin status of a representative locus is shown (bottom panel). f, Whole-genome bisulphite sequencing analysis on LTR7s. Comparison of the DNA methylation status of actively transcribing (highly active) and inactive elements in three different cell types: hiPSCs, hESCs and fibroblast. Average methylation levels are shown. Data from the ENCODE project and Epigenome Atlas (Supplementary Table 4).
Extended Data Figure 3 Pluripotency-associated transcription factors bind to HERVH.
a, All 5′ LTR7s of active HERVHs are associated with NANOG, while OCT4 is present in around 39. The plot combines the expression values of the 1,225 full-length HERVH (RNA-seq) with the fold-enrichment values of ChIP-seq data of OCT4 and NANOG in hESC_H1 (ref. 3). Each data point reflects a single full-length HERVH element. b, Motifs found to be significant in CLOVER and ROVER analyses. The four comparisons are active HERVH versus GC matched control sequence, HERVH flanked by LTR7 versus those flanked by LTR7C/Y, LTR7 itself against less active HERVH, and active HERVH versus active HERVK. We include only instances where the first two analyses both reported significance. Results for Tfcp2l1 (also called LBP9) are shown in red. c, EMSA confirms the binding of LBP9 to LTR7 sequence in vitro. Two different complexes (C#1 and C#2) were detected in the presence of nonspecific competitor (poly(dI-dC)). Complex 1 has lower stability (adding equal amount of competing oligonucleotide to the binding reaction doesn’t destroy it, but 100× excess does). Supershift is not detected with adding anti-LBP9 antibody, suggesting a lack of specificity, at least under our conditions. Complex 2 is resistant to being challenged with the competing oligonucleotide (100-fold excess), and supershifts with anti-LBP9 antibody, indicating specificity. From the low mobility we suspected that complex 2 is a large multimeric complex—this would also account for the modest but reproducible supershift. To explore the potentially multimeric nature of complex 2, we added anti-NANOG antibody. The supershift with anti-NANOG indicates that LBP9 binds LTR7 in a complex with NANOG. ESRG-oligonucleotide 50 nM (+); poly(dI-dC) 450 ng (+), 900 ng (++); anti-LBP9 5 μg (+), 10 μg (++); anti-NANOG 5 μg; competitor oligonucleotide 5 nM (+), 500 nM (++), 5,000 nM (+++); mutant oligonucleotide 50 nM; LBP9 ∼10 μg crude extract lysate in 20 μl total reaction volume. NS, nonspecific complex. For a list of oligonucleotide sequences, see Supplementary Table 1. d, Relative mRNA expression levels of HERVH correlates with pluripotency-associated transcription factors (OCT4, NANOG, and LBP9) during in vitro differentiation of hiPSCs. mRNA levels are normalized to GAPDH and relative to day 0. Error bars indicate s.d. from three independent cell cultures per time point.
Extended Data Figure 4 HERVH-driven transcription in hPSCs.
a, HERVH affects the neighbouring gene expression and produces HERVH-specific ‘chimaeric’ transcripts (RNA-seq reads which span HERVH and coding exons of neighbouring genes). Venn diagram shows the overlap between affected genes (see also Supplementary Tables 8 and 9). Examples of genes from each category are shown in boxes. b, Genes associated with HERVH function in stem cells with previously described gene functions. c, TSS distribution around HERVHs and the relationship between TSS identification and gene activity. CAGE data (from ENCODE) were analysed to identify TSS enriched on 5′ end active HERVHs. d, Expression heat map of 54 HERVH-derived lncRNAs in hPSCs and differentiated cells. Analysis of RNA-seq data sets as in Extended Data Fig. 1c. Data are displayed as log2 RPKM with high and low expression shown in red and blue, respectively. EB, embryoid body (data from this study). e, Knockdown effects of LBP9 and HERVH on the highest expressed lncRNAs in hPSCs (selected from the list presented in d). mRNA levels are normalized to GAPDH, and relative to shGFP expressing, undifferentiated hESC_H9. Fold-change values relative to shGFP knockdown are shown. Note that the knockdown effects of LBP9 and HERVH are highly similar. f, Alignment of top 22 hPSC-specific/HERVH-derived lncRNAs predict a conserved core domain (CD, referred as LTR7-CD). Certain CDs, embedded within lncRNAs, are annotated as exons and predicted to have limited coding potential (see also Supplementary Table 11). g, Heat map of potential RNA–protein interactions (predicted by CatRAPID54). lncRNAs were selected from Extended Data Fig. 4f if they were: (1) highly expressed in hESCs; (2) downregulated in HERVH knockdown; (3) enriched in nucleus. The Z-score describes the deviation of the studied RNA–protein interaction propensity from the ones based on randomized 100 RNAs against randomized 100 proteins (calculated by CatRAPID). The core domain of HERVH-derived lncRNAs is predicted to bind RNA-binding proteins, including pluripotency factors (for example, NANOG), and histone modifiers (for example, SET1A and SETDB1). High and low interaction potentials are shown in red and blue, respectively.
Extended Data Figure 5 LBP9/HERVH-driven transcription regulates pluripotency in hPSCs.
a, b, Characterization of hiPSC lines induced by OSKM+LBP9, OSKM+ESRG and OSKM+LTR7-CD by immunostaining (scale bar, 100 μm). a, Immunostaining for pluripotency markers. b, hiPSCs induced by OSKM+LBP9, OSKM+ESRG and OSKM+LTR7-CD can be differentiated into three germ layer lineages in vitro. c, Relative expression values of reprogramming-associated genes in HFF-1 are shown at different time points (RT–qPCR). Data normalized to GAPDH, and relative to day 0. Error bars indicate s.d. (n = 3 independent experiments with biological triplicates per experiment). d, Schematic representation of the regions of HERVH targeted by shRNA constructs shHERVH#3, shHERVH#4 and shHERVH#12. Predicted direct targets of shRNAs are shown in Supplementary Table 14. e, Validation of the shHERVH constructs. Stable, G418-resistant hESC-derived colonies express various shRNA constructs, targeting HERVH. Knockdown effect was monitored by qRT–PCR detecting either HERVH-gag or HERVH-pol levels. Data shown are representative of two independent experiments with biological triplicates per experiment. shHERVH#3, shHERVH#4 and shHERVH#12 knocked-down ∼80% of HERVH compared to the control shGFP. shHERVH#3, shHERVH#4 and shHERVH#12 (all shown in red) are also used in experiments presented in Fig. 3c–f. f, Representative immunostaining images showing reduction of pluripotency markers (OCT4, SOX2, SSEA4, and TRA-1-60) in both LBP9 and HERVH-depleted hESC_H9. shRNA against GFP was used as the control (shGFP). Scale bar, 100 μm. g, FACS analysis to determine the percentage of TRA-1-81+ cells after depletion of LBP9 or HERVH. Three different shRNAs were employed to independently target LBP9 and HERVH, respectively. Data are presented as mean and s.d. (n = 3 independent experiments with biological triplicates per experiment). h–j, Knockout of LBP9 in hESCs by the CRISPR/Cas9 technology. h, Experimental scheme to knockout LBP9 in hESCs using two guide RNAs (gRNAs), both targeting the second exon of LBP9. i, Analysis of LBP9 mutant hESC clones screened by genomic PCR. j, Sequence analysis of the TRA-1-81 sorted cells show that LBP9 mutants are found in differentiated (TRA-1-81−) but not in undifferentiated (TRA-1-81+) hESCs (representative samples). k, In contrast to human, Tfcp2l1 (mouse LBP9) depletion by shRNA does not affect self-renewal (left panel) in mouse ESCs in LIF/serum condition. Tfcp2l1-depleted mESCs were then differentiated into embryoid bodies (right panel), and endoderm and mesoderm markers were more expressed compared with shGFP mESC-derived embryoid bodies, indicating that Tfcp2l1-depleted mESCs have a bias to differentiate to endoderm and mesoderm (qRT–PCR analyses). Data are normalized to Gapdh, and relative to shGFP expressing, undifferentiated mESCs. Error bars indicate s.d. ND indicates undetectable. *P < 0.05, **P < 0.01, ***P < 0.001; t-test (n = 3 independent experiments with biological triplicates per experiment).
Extended Data Figure 6 ESRG is required for maintenance of human pluripotency.
a, Multi-alignment of ESRG putative open reading frame (ORF) from various primates. The ORF is intact in humans alone. All primate introns are shorter than the human one (which is 14,251 bp). The difference is dominantly accounted for by a single large insertion in the human sequence (around 2,000–7,500 bp) which comprises the bulk of the ESRG transcript (for alignment see Supplementary Data 1). b, Expression of ESRG during human embryogenesis24 and in hESC cultures3 (P, passage number). c–f, Characterization of the effects of ESRG depletion on hESC_H9. Note that knockdown of ESRG was performed by two different shRNA constructs, #4 and #5, respectively. shRNA against GFP served as a control. c, ESRG depletion compromises hESC self-renewal, indicated by the significant decline of the expression of pluripotency markers OCT4 and SSEA4. The expression of TRA-1-60 was also decreased, while SOX2 was unaffected. The representative images show immunostaining of pluripotency markers. Scale bar, 100 µm. d, FACS analysis of TRA-1-81 expression in ESRG-depleted hESCs by two different shRNA constructs. Data are shown as mean and s.d. (n = 3 independent experiments with biological triplicates per experiment). e, qRT–PCR analyses of ESRG knockdowns using selected markers (left, pluripotency; right, differentiation). Commitment to trophectoderm was the most apparent, characterized by the significant change in the expression of CDX2 in the ESRG-depleted cells. Data, representative of three independent experiments with biological triplicates per experiment, are normalized to GAPDH, and relative to shGFP expressing, undifferentiated hESCs (hESC_H9). Mean and s.d.; *P < 0.05, **P < 0.01, ***P < 0.001; t-test. f, Representative images of immunostaining showing expression of PAX6 (neuroectoderm) and CDX2 (trophectoderm) in ESRG-depleted hESCs_H9. Scale bar, 100 µm.
Extended Data Figure 7 The reporter assay.
a, Schematic of the reporter construct, pT2-LTR7-GFP#2, comprising an LTR7 region amplified from the ESRG locus, fused to GFP-polyA, and flanked by inverted terminal repeats (ITRs) of the SB transposon-based integration vector22. A reporter line was established by co-transfecting pT2-LTR7-GFP#2 with SB100X into HFF-1. GFP signal is detectable in both mouse and human transgenic ESCs. Representative pictures of pT2-LTR7-GFP#2-marked hESC_H9s and mESCs are shown. In the human case we show a FACS-sorted single colony. In mouse, as all cells homogeneously express GFP, we show multiple unsorted colonies. Scale bar, 100 μm. b, Multiple LTR7s responding to the fibroblast–iPSC transition are capable of driving the GFP reporter. Compared to the positive control 2 (pT2-LTR7-GFP#2), four additional responsive LTR7s (#3–6) amplified from different genomic loci were tested in the reporter assay (transfected into hiPSCs). The GFP signal of the five clones correlates to the RPKM values of the RNA-seq (not shown). Mock is a negative control transfected with the empty vector (pUC19). Percentage of GFP+ cells (green) and mean fluorescent intensity (black) are shown. Data were obtained from three independent experiments. Error bars indicate s.d.; **P < 0.01, t-test. c, Reporter assays to validate candidate transcription factors driving transcription from LTR7/HERVH. GFP signal is detectable in the fibroblast-derived reporter line by FACS, following forced expression of NANOG, LBP9, OCT4, KLF4, SOX2 and MYC constructs. Quantification was performed at days 2 and 7 post-transfection. Control was transfected with the empty vector (pUC19). Data were obtained from two independent experiments, *P < 0.05, **P < 0.01, *** P < 0.001; two-way ANOVA followed by Bonferroni test. A synergism between NANOG and LBP9 is indicated. d, Schematic representation of a reporter construct (pT2-LTR7-GFP#1: wild type, WT) and its mutated version (ΔLBP9), where the LBP9 motif was deleted; the constructs were transfected into hiPSCs. FACS quantification of the GFP signal derived from wild-type and motif-deleted cells. Percentage of GFP+ cells (green) and mean fluorescent intensity (black) are shown. Data were obtained from three independent experiments. Error bars indicate s.d.; t-test, *P < 0.05. e, pT2-LTR7-GFP#2 marked, mosaic, primed hPSC colonies in conventional hESC medium consist of cells expressing HERVH at various levels, but contain GFPhigh cell populations with mESC morphology (indicated by white arrowheads). Representative hiPSC (left panel) and hESC_H9 (right panel) colonies are shown. A GFPhigh cell population is magnified. Scale bar, 200 μm. f–h, FACS-sorted GFPhigh and GFPlow hESC_H9 cells were cultured in 2i/LIF, NHSM4 and 3iL3 conditions, respectively. f, g, Representative images of GFPhigh and GFPlow cells cultured in the different conditions at day 3. Scale bar, 200 μm. f, Morphology and GFP fluorescence of GFPhigh, 3D colonies were comparably maintained in the three different naive culture conditions, but not in primed culture conditions (KOSR and mTeSR1). g, Representative images show flat, GFP-negative colonies derived from GFPlow hESC_H9 cultured in either of the different culture conditions. h, Quantification by FACS of GFP-positive cells on day 6 of culturing in five media conditions: 2i/LIF, NHSM4, 3iL3, KOSR and mTeSR1. We cultured both GFPlow and GFPhigh cells before sorting. Longer-term culturing of GFPhigh naive cells is most compatible with 3iL3 culture condition (not shown). Percentage of GFPhigh cells, GFPlow cells (bright and pale green) and mean fluorescent intensity (black) are shown. KOSR, knockout serum replacement medium. Error bars, s.d.; n = 3 independent cell cultures, representative of two independent experiments. i, j, Heterogeneity of GFPhigh cells cultured in different conditions. i, The percentages of different hESC colonies derived from the same initial GFPhigh population in different culture conditions. 3D/GFPhigh, domed colony with strong GFP signal; 2D/GFPlow, flat colony with weak GFP signal; mosaic, colonies containing at least two cell types of GFPhigh and either GFPlow or GFP−; 3D/GFP−, domed colony without detectable GFP signal; 2D/GFP−, flat colony without detectable GFP signal. i, 388–462 colonies were characterized per culture condition, using fluorescence microscopy. j, qRT–PCR analysis of expression levels of core pluripotency-associated transcription factors in different colony types under the 2i/LIF condition. Total RNA isolated from 10–15 colonies per colony type was reverse transcribed for qPCR. Error bars indicate s.d. (n = 3, technical replicates).
Extended Data Figure 8 Characterization of LTR7-GFP-marked hPSCs.
a, Genetically labelled (pT2-LTR7-GFP#2) human naive-like hESC_H9 and hiPSCs can be maintained in 2i/LIF medium for a longer period of time (followed by passage number = P9, >30 days) by re-plating (every 4–5 days), and by occasional sorting for the GFP marker. For optimal long-term culturing conditions, see Extended Data Fig. 7h. b, Single-cell cloning efficiency of GFPhigh versus GFPlow hESC_H9. ALP-stained colonies were counted one week after plating 1,000 cells of a single cell suspension (with or without ROCK inhibitor (ROCKi)). Data were obtained from three independent experiments. Error bars indicate s.d., *P < 0.01, t-test. c, Both GFPhigh and GFPlow hESCs_H9 are immunostained by the indicated pluripotency markers (OCT4, SOX2, SSEA4). Scale bar, 100 μm. d, GFPhigh cells can be differentiated, and display the markers of the three germ layers. Scale bar, 100 μm. e, qRT–PCR analysis of pluripotency-associated transcription factors during in vitro differentiation of GFPhigh and GFPlow hESC_H9s. FACS-sorted GFPhigh and GFPlow cells were cultured in human 2i/LIF medium and in conventional hESC medium for 3 days, respectively, before differentiation was triggered. Error bars indicate s.d. (n = 3 independent experiments with biological triplicates per experiment), **P < 0.01, ***P < 0.001, t-test. f, FACS quantification of TRA-1-60-positive cells in differentiated GFPhigh and GFPlow cells. Error bars indicate s.d. (n = 3 independent experiments with biological triplicates per experiment), t-test for each time point, **P < 0.01, ***P < 0.001. g, Representative confocal image obtained after immunostaining for H3K27me3 on a chimaeric hESC_H9 colony. GFPhigh cells (green) are marked with lower density of H3K27m3 (red) than GFPlow and GFP− cells, indicating a higher histone methylation status in the absence of GFP. Scale bar, 20 μm. h, Global expression comparison between GFPhigh, GFP+ and GFPlow cells. Hierarchical clustering of the mean expression values of global gene expression using Spearman’s correlation (heat map). Biological replicates are shown. i, Mapping of the integration site of the pT2-LTR7-GFP#2 reporter in GFPhigh cells. The single copy of the reporter is integrated on chromosome 20 (red box) in a transcriptionally active area, marked by H3K36me3 and H3K79me2. j, Karyotype analysis result indicating the normal karyotype of hESC_H9 which were used in the present study.
Extended Data Figure 9 Transcription driven by HERVH defines naive-like state of hPSCs.
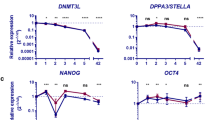
a, Expression of pluripotency-associated transcription factors in undifferentiated early (P0) and late passage (P10) hESCs24. At P10, n = 26; at P0, n = 8. t-test, *P < 0.05, ***P < 0.001. b, qRT–PCR analysis of pluripotency-associated transcription factors in undifferentiated early (P3) and late passage (P15) hiPSCs30, normalized to levels at P3. c, qRT–PCR analysis of pluripotency-associated transcription factors during in vitro differentiation of early (P3) and late passage (P15) hiPSCs. P, Passage number. t-test within each time period. d, Heat map showing differential HERVH transcription during human embryogenesis24 and in cultured hESCs3. The raw RNA-seq data downloaded from GEO (GSE36552) and ArrayExpress (E-MTAB-2031) were analysed to monitor HERVH expression. The rows represent the expression of 1,225 full-length HERVH loci. e, The average transcription of and number of active HERVHs during human embryogenesis and in cultured hESCs. f, Chromatin status comparison around full-length HERVHs between naive and primed hESC_H1 (ref. 3). While there are no differences in shared HERVH loci, which are transcribed in both naive and primed hESCs, the 5′ LTR of naive-specific HERVH loci are marked with H3K4me3. g, Heat map showing the comparison with mESC versus mouse epiblast stem cells (mEpiSCs53) of HERVH neighbour genes. Log2 fold change values of orthologous genes were subjected to hierarchical clustering (Pearson correlation, centroid linkage, k = 3). Genes selected and clustered as in h. h, The expression of neighbouring genes to HERVH in different human cell types, including GFPhigh, HERVH-depleted hPSCs, published naive hPSCs (naive(WIBR3)) and primed hESCs (reprimed(WIBR3))4. The heat map shows the comparison of row-normalized differential expression levels at log2 scale of fold changes of GFPhigh versus GFPlow, shHERVH versus shGFP, naive WIBR3 hESC versus primed and re-primed WIBR3 (GSE46872). Genes shown are those differentially expressed within every pairwise comparison (differential expression defined by log2 modular change >1, with FDR cutoff at 0.01). Isoform expression merged to single gene. Samples are represented in the order of euclidean distance and were clustered using Spearman’s correlation and centroid linkage. i, Scatter plot showing the differentially expressed genes between GFPhigh and GFPlow are negatively correlated with the ones between HERVH-depleted hESCs and wild-type hESCs. The enlisted genes are enriched in GFPhigh versus GFPlow and are specific to naive state (upper right), while genes downregulated by HERVH depletion are specific to primed hESCs or lineage commitment (lower). Red dots indicate differentially expressed genes. Representative clusters are shown. j, Gene ontology (GO) categories for downregulated genes in GFPhigh compared to GFPlow as well as naive hPSCs and mESCs versus primed cells4,53. k, Depletion of HERVH induced reduction of key transcription factors for naive hPSCs in the 2i/LIF medium. The representative images show the effects on GFPhigh cell morphologies upon depletion of HERVH. Scale bar, 100 μm. mRNA levels are normalized to GAPDH, and relative to shGFP expressing, undifferentiated hESC_H9. In b, c and k, error bars indicate s.d. (n = 3 independent experiments with biological triplicates per experiment), t-test, *P < 0.05, **P < 0.01, ***P < 0.001.
Extended Data Figure 10 HERVH drives a primate-specific naive pluripotency: a model.
a, The binding sites of transcription factors for naive pluripotency are clustered on HERVH. LBP9 is a modulator of the CP2 transcription-factor family28, and can form heteromeric, activator or repressor complexes with other family members, CP2, LBP1b, respectively. The activator complex interacts with OCT4 (ref. 16) and promotes pluripotency. In addition we provide evidence for the potential interaction of LBP9 and NANOG. Activated HERVHs generate numerous novel, stem-cell-specific alternative gene products. HERVH incorporates a set of regulatory lncRNAs into the network and defines novel pluripotency-associated gene products through alternative splicing (in conjunction with CHD1) or alternative non-AUG usage (in conjunction with other members of the CP2 family55). lncRNAs, some with a conserved domain (cruciform structure), interact with both pluripotency and chromatin modifying proteins (in green and blue). HERVH inhibits differentiation, while HERVH-derived products contribute to maintain pluripotency. LBP1b interacts with KRAB-associated protein 1 (KAP1, also called TRIM28), a repressor of ERVs during differentiation56. b, GFPhigh cells form dome-shaped colonies (3D), while GFPlow cells form flat (2D) colonies. Left: upregulated genes in GFPhigh cells include (1) naive transcription factors associated with HERVH (brown); (2) LTR7/HERVH driven novel isoforms of genes (*) and novel genes (for example, ESRG) (green); (3) naive transcription factors shared between mice and human (blue). Right: upregulated genes in GFPlow cells are associated with lineage commitment.
Supplementary information
Supplementary Information
This file contains a Supplementary Discussion and Supplementary References. (PDF 121 kb)
Supplementary Information
This file contains Supplementary Tables 1-16 and a Supplementary Table Guide. (ZIP 5560 kb)
Supplementary Data
This file contains an html rendering of alignment of the intron containing ESRG, as well as human ESRG, across multiple primates. (HTML 3888 kb)
Spatial structure visualization of the naïve state GFP(high) cells in a dome shaped hESC_H9 colony.
The colonies are genetically marked with GFP and immunostained with NANOG. Red, NANOG; green, GFP; blue, DAPI (nucleus); scare bar, 20 μM. Layer scanning was performed and images were taken using a Leica LSM710 point--‐scanning single photon confocal microscope. 3D image movies construction were created by Imaris Imaging Software (Bitplane). The colony shows mESC--‐like morphology (3D, multilayer). Note that high GFP fluorescence and NANOG staining appears in the same cells. (MP4 13347 kb)
Spatial structure visualization of the naïve state GFP(high) cells in a mosaic hESC_H9 colony.
The colonies are genetically marked with GFP and immunostained with NANOG. Red, NANOG; green, GFP; blue, DAPI (nucleus); scare bar, 20 μM. Layer scanning was performed and images were taken using a Leica LSM710 point--‐scanning single photon confocal microscope. 3D image movies construction were created by Imaris Imaging Software (Bitplane). The mosaic colony shows typical hESC morphology (2D, monolayer). (MP4 9231 kb)
Rights and permissions
About this article

Cite this article
Wang, J., Xie, G., Singh, M. et al. Primate-specific endogenous retrovirus-driven transcription defines naive-like stem cells. Nature 516, 405–409 (2014). https://doi.org/10.1038/nature13804
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature13804
This article is cited by
-
Ribosomal profiling of human endogenous retroviruses in healthy tissues
BMC Genomics (2024)
-
Selection on synonymous sites: the unwanted transcript hypothesis
Nature Reviews Genetics (2024)
-
Transposable elements activation triggers necroptosis in mouse embryonic stem cells
Cell Death & Disease (2023)
-
An old model with new insights: endogenous retroviruses drive the evolvement toward ASD susceptibility and hijack transcription machinery during development
Molecular Psychiatry (2023)
-
Crosstalk between RNA m6A and DNA methylation regulates transposable element chromatin activation and cell fate in human pluripotent stem cells
Nature Genetics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
