
Abstract
Animal transcriptomes are dynamic, with each cell type, tissue and organ system expressing an ensemble of transcript isoforms that give rise to substantial diversity. Here we have identified new genes, transcripts and proteins using poly(A)+ RNA sequencing from Drosophila melanogaster in cultured cell lines, dissected organ systems and under environmental perturbations. We found that a small set of mostly neural-specific genes has the potential to encode thousands of transcripts each through extensive alternative promoter usage and RNA splicing. The magnitudes of splicing changes are larger between tissues than between developmental stages, and most sex-specific splicing is gonad-specific. Gonads express hundreds of previously unknown coding and long non-coding RNAs (lncRNAs), some of which are antisense to protein-coding genes and produce short regulatory RNAs. Furthermore, previously identified pervasive intergenic transcription occurs primarily within newly identified introns. The fly transcriptome is substantially more complex than previously recognized, with this complexity arising from combinatorial usage of promoters, splice sites and polyadenylation sites.
Similar content being viewed by others


Main
Next-generation RNA sequencing (RNA-seq) has permitted the mapping of transcribed regions of the genomes of a variety of organisms1,2. These studies demonstrated that large fractions of metazoan genomes are transcribed, and they also catalogued individual elements of transcriptomes, including transcription start sites3, polyadenylation sites4,5, exons and introns6. However, the complexity of the transcriptome arises from the combinatorial incorporation of these elements into mature transcript isoforms. Studies that inferred transcript isoforms from short-read sequence data focused on a small subset of isoforms, filtered using stringent criteria7,8. Studies using complementary DNA (cDNA) or expressed sequence tag (EST) data to infer transcript isoforms have not had sufficient sampling depth to explore the diversity of RNA products at most genomic loci9. Although the human genome has been the focus of intensive manual annotation10, analysis of strand-specific RNA-seq data from human cell lines reveals over 100,000 splice junctions not incorporated into transcript models11. Thus, a large gap exists between genome annotations and the emerging transcriptomes observed in next-generation sequence data. In Drosophila, we previously described a non-strand-specific poly(A)+ RNA-seq analysis of a developmental time course through the life cycle6 and cap analysis of gene expression (CAGE) analysis of the embryo12, which discovered thousands of unannotated exons, introns and promoters, and expanded coverage of the genome by identified transcribed regions, but not all elements were incorporated into full-length transcript models. Here we describe an expansive poly(A)+ transcript set modelled by integrative analysis of transcription start sites (CAGE and 5′ rapid amplification of cDNA ends (RACE)), splice sites and exons (RNA-seq), and polyadenylation sites (3′ expressed sequence tags (ESTs), cDNAs and RNA-seq). We analysed poly(A)+ RNA data from a diverse set of developmental stages6, dissected organ systems and environmental perturbations; most of this data is new and strand-specific. Our data provide higher spatiotemporal resolution and allow for deeper exploration of the Drosophila transcriptome than was previously possible. Our analysis reveals a transcriptome of high complexity that is expressed in discrete, tissue- and condition-specific messenger RNA and lncRNA transcript isoforms that span most of the genome and provides valuable insights into metazoan biology.
A dense landscape of discrete poly(A)+ transcripts
To broadly sample the transcriptome, we performed strand-specific, paired-end sequencing of poly(A)+ RNA in biological duplicate from 29 dissected tissue samples including the nervous, digestive, reproductive, endocrine, epidermal and muscle organ systems of larvae, pupae and adults. To detect RNAs not observed under standard conditions, we sequenced poly(A)+ RNA in biological duplicate from 21 whole-animal samples treated with environmental perturbations. Adults were challenged with heat-shock, cold-shock, exposure to heavy metals (cadmium, copper and zinc), the drug caffeine or the herbicide paraquat. To determine whether exposing larvae resulted in RNA expression from previously unidentified genes, we treated them with heavy metals, caffeine, ethanol or rotenone. Finally, we sequenced poly(A)+ RNA from 21 previously described13 and three ovary-derived cell lines (Supplementary Methods). In total, we produced 12.4 billion strand-specific read pairs and over a terabase of sequence data, providing 44,000-fold coverage of the poly(A)+ transcriptome.
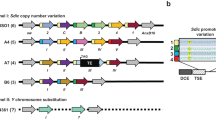
Reads were aligned to the Drosophila genome as described6, and full-length transcript models were assembled using our custom pipeline termed GRIT14, which uses RNA-seq, poly(A)+seq, CAGE, RACE12, ESTs15 and full-length cDNAs16 to generate gene and transcript models (Supplementary Methods). We integrated these models with our own and community manual curation data sets to obtain an annotation (Supplementary Information, section 12) consisting of 304,788 transcripts and 17,564 genes (Fig. 1a and Supplementary Fig. 1), of which 14,692 are protein-coding (Supplementary Data 1 and updates available at http://fruitfly.org). Ninety per cent of genes produce at most 10 transcript and five protein isoforms, whereas 1% of genes have highly complex patterns of alternative splicing, promoter usage and polyadenylation, and may each be processed into hundreds of transcripts (Fig. 1a, b). Our gene models span 72% of the euchromatin, an increase from 65% in FlyBase 5.12 (FB5.12), the reference annotation at the beginning of the modENCODE project (Supplementary Table 1 compares annotations in 2008–13). There were 64 euchromatic gene-free regions longer than 50 kb in FB5.12, and 25 remaining in FB5.45. Our annotation includes new gene models in each of these regions. Newly identified genes (1,468 total) are expressed in spatially and temporally restricted patterns (Supplementary Fig. 2), and 536 reside in previously uncharacterized gene-free regions. Others map to well-characterized regions, including the ovo locus, where we discovered a new ovary-specific, poly(A)+ transcript (Mgn94020, Supplementary Data 1 and 2), extending from the second promoter of ovo on the opposite strand and spanning 107 kb (Fig. 1c). Exons of 36 new genes overlap molecularly defined mutations with associated phenotypes (genome structure correction (GSC) P value ∼0.0002), indicating potential functions (Supplementary Table 2). For example, the lethal P-element insertions l(3)L3051 and l(3)L4111 (ref. 17) map to promoters of Mgn095159 and Mgn95009, respectively, indicating these may be essential genes. Nearly 60% of the intergenic transcription we previously reported6 is now incorporated into gene models.

a, Scatterplot showing the per gene correlation between number of proteins and number of transcripts. The genes Dscam and para are omitted as extreme outliers both encoding >10,000 unique proteins. b, Dystrophin (Dys) produces 72 transcripts and encodes 32 proteins. Highlighted is alternative splicing and polyadenylation at the 3′ end. CAGE (black), RNA-seq (tan, blue), splice junctions (shaded grey as a function of usage). c, An internal promoter of ovo is bidirectional in ovaries and produces a lncRNA (430 bp, red) bridging two gene deserts. CAGE (black), RNA-seq (pink), counts are read-depth (minus-strand given as negative).
Transcript diversity
Over half of spliced genes (7,412; 56%) encode two or more transcript isoforms with alternative first exons. Most of such genes produce alternative first exons through coordinated alternative splicing and promoter usage (59%, 4,389 genes, hypergeometric P value < 1 × 10−16); however, a substantial number of genes use one, but not both mechanisms (Fig. 2a). Only 1,058 spliced genes have alternative first exons that alter protein-encoding capacity and increase the complexity of the predicted proteome. Some genes, such as G protein β-subunit 13F (Gβ13F, Fig. 2b and Supplementary Fig. 3) have exceptionally complex 5′ UTRs, but encode a single protein.

a, Alternative first exons occur in two main configurations: multiple transcription start sites (TSS, pink) and multiple donor sites (DS, light blue). A subset of the genes in the multiple TSS category produce transcripts with different TSSs and shared donor sites (red), and a subset of the genes in the DS category produce transcripts with a shared TSS and different donor sites (blue). Some genes in the multiple TSS category directly affect the encoded protein (maroon), and similarly for DS (dark blue). The overlap of configurations is radially proportional (units indicate percentage of all spliced genes). b, Poly(A)+ testes (blue) and central nervous system (CNS) (orange) stranded RNA-seq of Gβ13F showing complex processing and splicing of the 5′ UTR. An expansion of the 5′ UTR showing some of the complexity. Transcription of the gene initiates from one of three different promoters (green arrows) terminates at one of ten possible poly(A)+ addition sites (from adult head poly(A)+seq, red) and generates 235 transcripts. The first exon has two alternative splice acceptors that splice to one of eleven different donor sites. Only five donor sites are shown owing to the proximity of splice sites. Four splice donors are represented by the single red line differing by 12, 5 and 19 bp, respectively. Three splice donors are represented by the single green line differing by 12 and 11 bp. Two splice donors are represented by the single purple line differing by 7 bp. These splice variants are combined with four proximal internal splices (Supplementary Fig. 3a) to generate the full complement of transcripts. c, Intron retention rates (Ψ) across the gene body. The genome-wide mean lengths of exons and introns are connected by red parabolic arcs, which illustrate the upper and lower quartiles of intron retention (across all samples) for introns retained at or above 20 Ψ in at least one sample.
We measured splicing efficiency using the ‘per cent spliced in’ (Ψ) index—the fraction of isoforms that contain the particular exon6. Introns flanked by coding sequence are retained at an average Ψ = 0.7, whereas introns flanked by non-coding sequence are retained > fivefold more often, with an average Ψ = 3.8 (P < 1× 10−16 subsampling/two-sample t-test), and is most frequent in 5′ UTRs (mean Ψ = 5.1, Fig. 2c).
Despite the depth of our RNA-seq, these data show that 42% of genes encode only a single transcript isoform, and 55% encode a single protein isoform (Supplementary Methods). In mammals, it has been estimated that 95% of genes produce multiple transcript isoforms18,19, (estimates for protein-coding capacity have not been reported).
The majority of transcriptome complexity is attributable to forty-seven genes that have the capacity to encode >1,000 transcript isoforms each (Supplementary Table 3), and account for 50% of all transcripts (Fig. 3a). Furthermore, 27% of transcripts encoded by these genes were detected exclusively in samples enriched for neuronal tissue, and another 56% only in the embryo (83% total). To determine their tissue specificities we conducted embryonic in situ expression assays (Fig. 3b) and found that 18 of 35 are detected only in neural tissue (51% compared with 10% genome-wide, hypergeometric P value < 1 × 10−16, Supplementary Table 4). Of these genes, 48% have 3′ UTR extensions in embryonic neural tissue20 (5% genome-wide, P < 1× 10−16). Furthermore, 44% are targets of RNA editing (4% genome-wide6, P < 1 × 10−16, with 18 of 21 validated21), and 21% have 3′ UTR extensions and RNA editing sites (10 of 65 genome-wide, P < 1× 10−100). The capacity to encode thousands of transcripts is largely specific to the nervous system and coincides with other classes of rare, neural-specific RNA processing.

a, A small minority of genes (47, 0.2%) encode most transcripts. b, In situ RNA staining of constitutive exons of four genes with highly complex splicing patterns in the embryo. Syncrip (Syp), CAP , retinal degeneration A (rdgA) and GluClα show specific late embryonic neural expression in the ventral midline neurons; dorsal/lateral and ventral sensory complexes; Bolwig’s organ or larval eye; and central nervous system, respectively.
Tissue- and sex-specific splicing
To examine the dynamics of splicing, we calculated switch scores or ΔΨ, for each splicing event by comparing the maximal and minimal Ψ values across all samples, and in subsets including just the developmental and tissue samples. In contrast to the median Ψ values, the distribution of ΔΨ values is strikingly different between the developmental and tissue samples. Among the developmental samples, 38% of events have a ΔΨ ≥ 50%, whereas between the tissue samples 63% of events have a ΔΨ ≥ 50%. This difference is even more pronounced at higher ΔΨ thresholds—only 6% of events have a ΔΨ ≥ 80% between the developmental samples, whereas 31% of events have a ΔΨ ≥ 80% between the tissue samples. Thus, most splicing events are highly tissue-specific. Of the 17,447 alternative splicing events analysed (Supplementary Information, section 19), we find that 56.6% changed significantly (ΔΨ > 20%, Bayes factor >20). Clustering revealed groups of splicing events that are co-ordinately regulated in a tissue-specific manner. For example, 1,147 splicing events are specifically included in heads and excluded in testes or ovaries, whereas 797 splicing events are excluded in heads but included in testes or ovaries (Fig. 4a).

a, Clusters of tissue-specific splicing events. The scale bar indicates z-scores of Ψ. Adult mated male (AdMM), adult mated female (AdMF) and adult virgin female (AdVF) heads are from 1-, 4- and 20-day-old animals, respectively. Testes are from 4-day-old adult males, and ovaries are from mated and virgin 4-day-old adult females. b, Sex-specific splicing events in whole animals are primarily testes- or ovary-specific splicing events. Adult male (AdM) and adult female (AdF) animals are 1, 5 and 30 days old. Accessory glands were dissected from 4-day-old adult males. The RNA-seq columns from heads, testes and ovaries are as described in a.
We identified hundreds of sex-specific splicing events from adult male and female RNA-seq data6. To further explore sex-specific splicing, we compared the splicing patterns in male and female heads enriched for brain tissues. There were striking differences in gene expression levels, however, only seven splicing events were consistently differentially spliced at each time point after eclosion (average ΔΨ > 20%), and these largely corresponded to genes in the known sex-determination pathway (Supplementary Information, section 19A). We find few examples of head sex-specific splicing. This is in contrast to previous studies, which have come to conflicting conclusions and used either microarrays analysing only a subset of splicing events or single read 36-bp RNA-seq22,23 with an order of magnitude fewer reads24.
We identified 575 alternative splicing events that are differentially spliced in whole male and female animals (ΔΨ > 20%) and analysed the tissue-specific splicing patterns of each event (Fig. 4b). We found that 186 of the 321 male-biased splicing events were most strongly included in testes or accessory glands, and 157 of 254 female-biased exons were ovary-enriched. Consistent with the extensive transcriptional differences observed in testes compared to other tissues, the genes containing male-specific exons are enriched in functions related to transcription. In contrast, the female-specific exon containing genes are enriched in functions involved in signalling and splicing ((http://reactome.org)25, Supplementary Table 6). Together, these results indicate that the majority of sex-specific splicing is due to tissue-specific splicing in tissues present only in males or females.
Long non-coding RNAs
A growing set of candidate long non-coding RNAs (lncRNAs) have been identified in Drosophila6,26,27. In FB5.45 there were 392 annotated lncRNAs, and it has been suggested that as many as 1,119 lncRNAs may be transcribed in the fly28. However, this number was based on transcribed regions, not transcript models, and used non-stranded RNA-seq data28. We find 3,880 genes produce transcripts with ORFs encoding fewer than 100 amino acids. Of these, 795 encode conserved proteins (Methods) longer than 20 amino acids. For example, a single exon gene on the opposite strand and in the last intron of the early developmental growth factor spätzle encodes a 42-amino-acid protein that is highly conserved across all sequenced Drosophila species. We identified 1,875 candidate lncRNA genes producing 3,085 transcripts, 2,990 of which have no overlap with protein-coding genes on the same strand (Supplementary Data 2). Some of these putative lncRNAs may encode short polypeptides, for example, the gene tarsal-less encodes three 11-amino-acid ORFs with important developmental functions29. We determined protein conservation scores for each ORF between 20 and 100 amino acids (Supplementary Table 6). Of the 1,119 predicted lncRNAs28, we provide full-length transcript models for 246 transcribed loci; the remainder were expressed at levels beneath thresholds used in this study. This is not surprising, the expression patterns of lncRNAs are more restricted than those of protein-coding genes: the average lncRNA is expressed (bases per kilobase per million mapped bases6 (BPKM) > 1) in 1.5 developmental and 3.2 tissue samples, compared to 6.6 and 17 for protein-coding genes, respectively. Many lncRNAs (563 or 30%) have peak expression in testes, and 125 are detectable only in testes. Similarly restricted expression patterns have been reported for lncRNAs in humans and other mammals30,31.
Interestingly, all newly annotated genes overlapping molecularly defined mutations with phenotypes are lncRNAs (Supplementary Table 2). For instance, the mutation D114.3 is a regulatory allele of spineless (ss) that maps 4 kb upstream of ss32 and within the promoter of Mgn4221. Similarly, Mgn00541 corresponds to a described, but unannotated 2.0 kb transcript overlapping the regulatory mutant allele ci57 of cubitus interruptus33. It remains to be determined whether these mutations are a result of the loss-of-function of newly annotated transcripts or cis-acting regulatory elements (for example, enhancers) or both.
Antisense transcription
Drosophila antisense transcription has been reported34, but the catalogue of antisense transcription has been largely limited to overlapping mRNAs transcribed on opposite strands. We identify non-coding antisense transcript models for 402 lncRNA loci that are antisense to mRNA transcripts of 422 protein-coding genes (for example, prd, Fig. 5a), and 36 lncRNAs form ‘sense-antisense gene-chains’ overlapping more than one protein-coding locus, as observed in mammals30,35. In Drosophila, 21% of lncRNAs are antisense to mRNAs, whereas in human 15% of annotated lncRNAs are antisense to mRNAs (GENCODE v.10). We assembled antisense transcript models for 5,057 genes (29%, compared to previous estimates of 15%34). For 67% of these loci, antisense expression is observable in at least one cell line, indicating that sense/antisense transcripts may be present in the same cells. LncRNA-mediated antisense accounts for a small minority of antisense transcription: 94% of antisense loci correspond to overlapping protein-coding mRNAs transcribed on opposite strands, and of these, 323 loci (667 genes) share overlapping CDSs. The majority of antisense is due to overlapping UTRs: 1,389 genes have overlapping 5′ UTRs (divergent transcription), 3,430 have overlapping 3′ UTRs (convergent transcription), and 540 have both, meaning that, as with many lncRNAs, they form gene-chains across contiguously transcribed regions. A subset of antisense gene-pairs overlap almost completely (>90%), which we term reciprocal transcription. There are 13 such loci (Supplementary Fig. 5) and seven are male-specific (none are female-specific).

a, 5′/5′ overlapping bidirectional antisense transcription at the prd locus. Short RNA sequencing does not reveal substantial siRNA (that is, 21-nt-dominant small RNA) signal in this region (data not shown). b, A 5′/5′ antisense region that produces substantial small RNA signal on both strands. nt, nucleotide.
The mRNA/lncRNA sense-antisense pairs tend to be more positively correlated in their expression than mRNA/mRNA pairs, (mean r = 0.16 compared with 0.13, Kolmogorov–Smirnov (KS) two-sample one-sided test P < 10−9), and although this mean effect is subtle, the trend is clearly visible in the quantiles (95th percentile lncRNA/mRNA 0.729 versus mRNA/mRNA 0.634, Supplementary Fig. 6a). This effect is stronger when the analysis is restricted to cell line samples (Supplementary Fig. 6b).
Even in homogenous cell cultures, evidence for sense-antisense transcription does not guarantee that both transcripts exist within individual cells: transcription could originate from exclusive events occurring in different cells. Cis-natural antisense transcripts (cis-NATs) are a substantial source of endogenous siRNAs36, and their existence directly reflects the existence of precursor dsRNA. Cis-NAT-siRNA production typically involves convergent transcription units that overlap on their 3′ ends, but other documented loci generate siRNAs across internal exons, introns or 5′ UTRs37,38,39. Analysis of head, ovary and testis RNAs showed that 328 unique sense/antisense gene pair regions generated 21-nucleotide RNAs indicative of siRNA production (Supplementary Table 8), and these were significantly enriched (Supplementary Fig. 7a, Supplementary Methods) for pairs showing positively correlated expression between sense and antisense levels across tissues (P = 2 × 10−5), embryo developmental stages (P = 4 × 10−3), conditions (P = 9 × 10−4) and across all samples (P = 3 × 10−5). The tissue distribution of these cis-NAT-siRNAs showed a bias for testis expression (Supplementary Fig. 7b), with fourfold greater number relative to ovaries (P = 2 × 10−17, binomial test) and sevenfold relative to heads (P = 4 × 10−25) and expression levels of siRNAs were substantially higher in testes than other tissues (Supplementary Fig. 7c).
Over 80% of cis-NAT-siRNAs were derived from 3′-convergent gene pairs. Abundant siRNAs emanate from an overlap of the gryzun and CG14967 3′ UTRs (Supplementary Fig. 5). The remainder were distributed amongst CDSs, introns and 5′ UTRs. We identified abundant testis-enriched siRNA production from a 5′-divergent overlap of Cyt-c-d and CG31808 (Fig. 5b) and from the entire CDS of dUTPase and its antisense non-coding transcript Mgn99994.
Transcriptional effects of environmental stress
Whole-animal perturbations each exhibited condition-specific effects, for example, the metallothionein genes were induced by heavy metals (Fig. 6a), but not by other treatments (Supplementary Table 9). The genome-wide transcriptional response to cadmium (Cd) exposure involves small changes in expression level in thousands of genes (48 h after exposure), but only a small group of genes change > 20-fold, and this group includes six lncRNAs (the third most strongly induced gene is CR44138, Fig. 6a, Supplementary Fig. 8a). Four newly modelled lncRNAs are differentially expressed (1% false discovery rate (FDR)) in at least one treatment, and constitute newly described eco-responsive genes. Furthermore, 57 genes and 5,259 transcripts (of 811 genes) were detected exclusively in these treatment samples. Although no two perturbations revealed identical transcriptional landscapes, we find a homogeneous response to environmental stressors (Fig. 6b, Supplementary Fig. 8b). The direction of regulation for most genes is consistent across all treatments; very few are upregulated in one condition and downregulated in another. Classes of strongly upregulated genes included those annotated with the GO term “Response to Stimulus, GO:0050896” (most enriched, P value < 1 × 10−16, Supplementary Fig. 8c), and those that encode lysozymes (> tenfold), cytochrome P450s, and mitochrondrial components mt:ATPase6, mt:CoI, mt:CoIII (> fivefold). Genes encoding egg-shell, yolk and seminal fluid proteins are strongly downregulated in response to every treatment except ‘cold2’ and ‘heat shock’ (Supplementary Fig. 8d). For these two stressors, samples were collected 30 min after exposure, corresponding to an ‘early response test’ showing suppression of germ cell production is not immediate.

Adults were treated with caffeine (Cf), Cd, Cu, Zn, cold, heat or paraquat (PQ). a, A genome-wide map of genes that are up- or downregulated as a function of Cd treatment. Labelled genes are those that showed a 20-fold (<10% FDR) change in response (linear scale). Genes highlighted in red are those identified in larvae50. Some genes are omitted for readability, the complete figure and list of omitted genes are given in Supplementary Fig. 8a. b, Heat map showing the fold change of genes with a FDR < 10% (differential expression) in at least one sample (log2 scale). PL, pre-lethal.
Discussion
Most transcriptional complexity in Drosophila occurs in tissues of the nervous system, and particularly in the functionally differentiating central and peripheral nervous systems. A subset of ultra-complex genes encodes more than half of detected transcript isoforms and these are dramatically enriched for RNA editing events and 3′ UTR extensions, both phenomena largely specific to the nervous system. Our study indicates that the total information output of an animal transcriptome may be heavily weighted by the needs of the developing nervous system.
The improved depth of sampling and spatiotemporal resolution resulted in the identification of more than 1,200 new genes not discovered in our previous study of Drosophila development6. A large fraction of the new genes are testes-specific, and many of these are antisense RNAs, as previously described in mammals30. Some new lncRNAs, such as Mgn94020 (Fig. 1), form sense/antisense gene-chains that bring distant protein-coding genes into transcriptional relationships, another phenomenon previously described only in mammals40. Whenever Mgn94020 is detectably transcribed, the genes on the opposite strand in its introns are not, indicating that its transcription may serve a regulatory function independent of the RNA transcribed. The presence of short RNAs at many regions of antisense transcription indicates that sense and antisense transcripts are present in the same cells at the same times. Many of these Drosophila antisense transcripts correspond to ‘positionally equivalent’30 antisense transcripts in human. In the two species we found antisense lncRNAs opposite to orthologous protein-coding genes. The apparent positional equivalence of fly and human antisense transcription at genes like Monocarboxylate transporter 1 (MCT1), even-skipped (EVX1), CTCF (CTCF), Adenosine receptor (ADORA2A), and many others10,31 across 600 million years of evolution suggests a conserved regulatory mechanism basal to sexual reproduction in metazoans.
Perturbation experiments identified new genes and transcripts, but perhaps more importantly, a general response to stress that is broader than the heat shock pathway. A similar study conducted on marsh fishes in the wake of the Deepwater Horizon incident in the Gulf of Mexico41 demonstrated that the killifish response to chronic hydrocarbon exposure included induction of lyzosome genes, P450 cytochromes and mitochondrial components, and the downregulation of genes encoding eggshell and yolk proteins41. This overlap of expressional responses by gene families across phyla suggests a conserved metazoan stress response involving enhanced metabolism and the suppression of genes involved in reproduction.
We defined an extensive catalogue of putative lncRNAs. However, many genes are known to encode poorly conserved, short polypeptides, including genes specific to the male gonad and accessory gland. Analysis of ribosome profiling initially indicated that a number of mammalian lncRNAs may be translated42, but this observation has been difficult to validate by proteomics43, and further analysis has suggested that although lncRNAs have signatures of ribosome occupancy, they are not translated44. Therefore, while we refer to these RNAs as ‘non-coding’, additional data are needed to determine if they produce small polypeptides.
The biological consequences of many of the phenomena reported here, including the observation that many genes encoding RNA binding proteins exhibit extraordinary splicing complexity, often within their 5′ UTRs, require further study. The splicing factor pUf68 encodes more than 100 alternatively spliced 5′ UTR variants, but encodes a single protein. The idea that splicing factors may regulate one another to generate complex patterns of splicing is consistent with recent computational models45. More generally, the role of complex splicing in the adult and developing nervous system is unclear. To answer the questions that come with increasingly complete transcriptomes in higher organisms, it will be necessary to study gene regulation downstream of transcription initiation, including the regulation of splicing, localization and translation.
Methods Summary
Animal staging, collection and RNA extraction
Tissues were dissected from Oregon R larval, pupal and adult staged animals synchronized with appropriate age indicators. Pupal and adult animals were treated with a number of environmental stresses. RNA was isolated using TRIzol (Invitrogen), treated with DNase and purified on a RNAeasy column (Qiagen). Poly(A)+ RNA was prepared from an aliquot of each total RNA sample using an Oligotex kit (Qiagen).
RNA-seq
Libraries were generated and sequenced on an Illumina Genome Analyzer IIx or HiSeq 2000 using paired-end chemistry and 76-bp or 100-bp cycles. The 454 sequencing used poly(A)+ RNA from Oregon R adult males and females and mixed-staged y1 cn1bw1 sp1. embryos. Sequences are available from the Short Read Archive (Accession numbers available in Supplementary Table 10) and the modENCODE website (http://www.modencode.org/, Supplementary Table 10). CAGE46 was sequenced on an Illumina Genome Analyzer IIx with 36-bp reads. Poly(A)+seq was generated using a custom protocol (Supplementary Methods).
Analysis
RNA-seq, CAGE and poly(A)+ reads were mapped and filtered12. GRIT was used to identify transcript models14. Expression levels for genes and exons were computed in BPKM6. GSC P values were computed47. Ψ values were calculated with MISO48. Differential expression analysis was conducted with a custom method (Supplementary Methods) and with DEseq49. RPS-BLAST was used to conduct the conserved domain search with version v3.08 of the NCBI Conserved Domains Database (CDD) (Supplementary Methods). Orthology analysis between human and fly was conducted using DIOPT (http://www.flyrnai.org/cgi-bin/DRSC_orthologs.pl). Phenotypic alleles were downloaded from FlyBase r5.50, and were selected as any allele localized to the genome with a disease phenotype.
Accession codes
Data deposits
Sequences are available from the Short Read Archive and the modENCODE website, a list of accession numbers is given in Supplementary Table 10.
References
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods 5, 621–628 (2008)
Nagalakshmi, U. et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320, 1344–1349 (2008)
Takahashi, H., Kato, S., Murata, M. & Carninci, P. CAGE (cap analysis of gene expression): a protocol for the detection of promoter and transcriptional networks. Methods Mol. Biol. 786, 181–200 (2012)
Mangone, M. et al. The landscape of C. elegans 3′UTRs. Science 329, 432–435 (2010)
Jan, C. H., Friedman, R. C., Ruby, J. G. & Bartel, D. P. Formation, regulation and evolution of Caenorhabditis elegans 3′UTRs. Nature 469, 97–101 (2011)
Graveley, B. R. et al. The developmental transcriptome of Drosophila melanogaster. Nature 471, 473–479 (2011)
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols 7, 562–578 (2012)
Collins, J. E., White, S., Searle, S. M. & Stemple, D. L. Incorporating RNA-seq data into the zebrafish Ensembl genebuild. Genome Res. 22, 2067–2078 (2012)
Carninci, P. et al. Targeting a complex transcriptome: the construction of the mouse full-length cDNA encyclopedia. Genome Res. 13, 1273–1289 (2003)
Harrow, J. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012)
Djebali, S. et al. Landscape of transcription in human cells. Nature 489, 101–108 (2012)
Hoskins, R. A. et al. Genome-wide analysis of promoter architecture in Drosophila melanogaster. Genome Res. 21, 182–192 (2011)
Cherbas, L. The transcriptional diversity of 25 Drosophila cell lines. Genome Res. 21, 301–314 (2011)
Boley, N. et al. Genome guided transcript construction from integrative analysis of RNA sequence data. Nature Biotechnol. http://dx.doi.org/10.1038/nbt.2850 (2014)
Celniker, S. E. & Rubin, G. M. The Drosophila melanogaster genome. Annu. Rev. Genomics Hum. Genet. 4, 89–117 (2003)
Stapleton, M. et al. The Drosophila gene collection: identification of putative full-length cDNAs for 70% of D. melanogaster genes. Genome Res. 12 1294–1300 (2002) 2
Spradling, A. C. et al. The Berkeley Drosophila Genome Project gene disruption project: single P-element insertions mutating 25% of vital Drosophila genes. Genetics 153, 135–177 (1999)
Wang, E. T. et al. Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476 (2008)
Pan, Q., Shai, O., Lee, L. J., Frey, B. J. & Blencowe, B. J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nature Genet. 40, 1413–1415 (2008)
Smibert, P. et al. Global patterns of tissue-specific alternative polyadenylation in Drosophila. Cell Rep. 1, 277–289 (2012)
St Laurent, G. et al. Genome-wide analysis of A-to-I RNA editing by single-molecule sequencing in Drosophila. Nature Struct. Mol. Biol. 20, 1333–1339 (2013)
Telonis-Scott, M., Kopp, A., Wayne, M. L., Nuzhdin, S. V. & McIntyre, L. M. Sex-specific splicing in Drosophila: widespread occurrence, tissue specificity and evolutionary conservation. Genetics 181, 421–434 (2009)
Hartmann, B. et al. Distinct regulatory programs establish widespread sex-specific alternative splicing in Drosophila melanogaster. RNA 17, 453–468 (2011)
Chang, P. L., Dunham, J. P., Nuzhdin, S. V. & Arbeitman, M. N. Somatic sex-specific transcriptome differences in Drosophila revealed by whole transcriptome sequencing. BMC Genomics 12, 364 (2011)
Matthews, L. et al. Reactome knowledgebase of human biological pathways and processes. Nucleic Acids Res. 37, D619–D622 (2009)
Lipshitz, H. D., Peattie, D. A. & Hogness, D. S. Novel transcripts from the Ultrabithorax domain of the bithorax complex. Genes Dev. 1, 307–322 (1987)
Tupy, J. L. et al. Identification of putative noncoding polyadenylated transcripts in Drosophila melanogaster. Proc. Natl Acad. Sci. USA 102, 5495–5500 (2005)
Young, R. S. et al. Identification and properties of 1,119 candidate lincRNA loci in the Drosophila melanogaster genome. Genome Biol. Evol. 4, 427–442 (2012)
Kondo, T. et al. Small peptide regulators of actin-based cell morphogenesis encoded by a polycistronic mRNA. Nature Cell Biol. 9, 660–665 (2007)
Katayama, S. et al. Antisense transcription in the mammalian transcriptome. Science 309, 1564–1566 (2005)
Derrien, T. et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789 (2012)
Duncan, D. M., Burgess, E. A. & Duncan, I. Control of distal antennal identity and tarsal development in Drosophila by spineless-aristapedia, a homolog of the mammalian dioxin receptor. Genes Dev. 12, 1290–1303 (1998)
Schwartz, C., Locke, J., Nishida, C. & Kornberg, T. B. Analysis of cubitus interruptus regulation in Drosophila embryos and imaginal disks. Development 121, 1625–1635 (1995)
Misra, S. et al. Annotation of the Drosophila melanogaster euchromatic genome: a systematic review. Genome Biology 3, research0083 (2002)
Lipovich, L. et al. Activity-dependent human brain coding/noncoding gene regulatory networks. Genetics 192, 1133–1148 (2012)
Okamura, K. & Lai, E. C. Endogenous small interfering RNAs in animals. Nature Rev. Mol. Cell Biol. 9, 673–678 (2008)
Okamura, K., Balla, S., Martin, R., Liu, N. & Lai, E. C. Two distinct mechanisms generate endogenous siRNAs from bidirectional transcription in Drosophila melanogaster. Nature Struct. Mol. Biol. 15, 581–590 (2008)
Czech, B. et al. An endogenous small interfering RNA pathway in Drosophila. Nature 453, 798–802 (2008)
Ghildiyal, M. et al. Endogenous siRNAs derived from transposons and mRNAs in Drosophila somatic cells. Science 320, 1077–1081 (2008)
Engström, P. G. et al. Complex loci in human and mouse genomes. PLoS Genet. 2, e47 (2006)
Whitehead, A. et al. Genomic and physiological footprint of the Deepwater Horizon oil spill on resident marsh fishes. Proc. Natl Acad. Sci. USA 109, 20298–20302 (2012)
Ingolia, N. T., Ghaemmaghami, S., Newman, J. R. & Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223 (2009)
Bánfai, B. et al. Long noncoding RNAs are rarely translated in two human cell lines. Genome Res. 22, 1646–1657 (2012)
Guttman, M., Russell, P., Ingolia, N. T., Weissman, J. S. & Lander, E. S. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell 154, 240–251 (2013)
Huelga, S. C. et al. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep. 1, 167–178 (2012)
Takahashi, H., Lassmann, T., Murata, M. & Carninci, P. 5′ end-centered expression profiling using cap-analysis gene expression and next-generation sequencing. Nature Protocols 7, 542–561 (2012)
Bickel, P. J., Boley, N., Brown, J. B., Huang, H. & Zhang, N. R. Subsampling methods for genomic inference. Ann. Appl. Stat. 4, 1660–1697 (2010)
Katz, Y., Wang, E. T., Airoldi, E. M. & Burge, C. B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nature Methods 7, 1009–1015 (2010)
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010)
Yepiskoposyan, H. et al. Transcriptome response to heavy metal stress in Drosophila reveals a new zinc transporter that confers resistance to zinc. Nucleic Acids Res. 34, 4866–4877 (2006)
Acknowledgements
We thank the members of the modENCODE transcription consortium, especially J. Landolin and J. Sandler for their early contributions to these studies. We also thank A. Kundaje and H. Huang for helpful discussions. This work was funded by a contract from the National Human Genome Research Institute modENCODE Project, contract U01 HG004271 and U54 HG006944, to S.E.C. (principal investigator) and P.C., T.R.G., R.A.H. and B.R.G. (co-principal investigators) with additional support from R01 GM076655 (S.E.C.) both under Department of Energy contract no. DE-AC02-05CH11231. J.B.B.’s work was supported by NHGRI K99 HG006698. Work in P.J.B.’s group was supported by the modENCODE DAC sub-award 5710003102, 1U01HG007031-01 and the ENCODE DAC 5U01HG004695-04. Work in Bloomington was supported in part by the Indiana METACyt Initiative of Indiana University, funded by an award from the Lilly Endowment. Work in E.C.L.’s group was supported by U01-HG004261 and RC2-HG005639.
Author information
Authors and Affiliations
Contributions
J.A., T.R.G., B.R.G., R.A.H., T.C.K. and S.E.C. designed the project. J.A., P.Ch., T.R.G., B.R.G., R.A.H., J.B.B., B.O. and S.E.C. managed the project. R.E. designed treatment protocols and prepared biological samples. T.C.K., J.A. and L.C. oversaw biological sample production. B.D.E., D.M. and J.R. prepared biological samples. D.Z. and B.E. prepared RNA samples. J.A. oversaw RNA sample production. G.E.M., S.O. and L.Y. prepared Illumina RNA-seq libraries. A.M.S. prepared CAGE libraries. P.Ca. oversaw production of CAGE libraries. C.A.D., G.E.M., S.O., L.Y., S.P. and K.H.W. performed Illumina sequencing. B.R.G. and S.E.C. managed Illumina sequencing production. R.A.H. conceived the poly(A)+seq method. R.W. and R.A.H. developed the poly(A)+seq protocol and produced the libraries. K.M. performed 454 sequencing. C.Y., S.P. and K.H.W. performed cDNA library screens and full-insert cDNA sequencing. S.E.C. oversaw cDNA production. E.F. and N.B. installed and administered computer infrastructure for data storage and analysis. J.B.B., N.B., M.H.S., M.O.D., B.W.B., D.S., J.W.C., S.S., J.W., A.A.S., N.P., E.C.L., P.J.B. and B.R.G. developed analysis methods. J.B.B., N.B., M.H.S., M.O.D., B.W.B., A.S.H., E.F., R.A.H., S.S., D.S., L.C., G.R., J.H., J.W., A.A.S., E.C.L., K.H.W., B.R.G. and S.E.C. analysed data. N.B., J.B.B. M.H.S., K.H.W. and S.E.C. generated annotations. D.S. and B.O. analysed species validation data. S.S., J.W. and E.C.L. analysed 3′ UTR and antisense data. A.S.H., E.F. and S.E.C. analysed image data. M.H.S. analysed proteomics data. M.H.S., S.S., D.S., B.O., E.C.L., T.C.K., R.E., R.A.H. and P.Ch. contributed to the text. A.S.H. assisted with manuscript preparation. J.B.B., B.R.G. and S.E.C. wrote the paper with input from all authors. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
This file contains Supplementary Text and References, a guide to the Supplementary Tables and Supplementary Data files and Supplementary Figures 1-11 – see the contents page for details. (PDF 2385 kb)
Supplementary Tables
This zipped file contains Supplementary Tables 1-10 - see Supplementary Information document p.13 for more details. (ZIP 2098 kb)
Supplementary Data
This zipped file contains Supplementary Data sets 1-6 - see Supplementary Information document p.13 for more details. (ZIP 19021 kb)
Supplementary Data
This zipped file contains Supplementary Data sets 7-9 - see Supplementary Information document p.13 for more details. (ZIP 28618 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-Non-Commercial-ShareAlike 3.0 Unported licence. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-sa/3.0/.
About this article
Cite this article
Brown, J., Boley, N., Eisman, R. et al. Diversity and dynamics of the Drosophila transcriptome. Nature 512, 393–399 (2014). https://doi.org/10.1038/nature12962
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature12962
This article is cited by
-
Genome-wide association in Drosophila identifies a role for Piezo and Proc-R in sleep latency
Scientific Reports (2024)
-
Slik maintains tissue homeostasis by preventing JNK-mediated apoptosis
Cell Division (2023)
-
Mapping splice QTLs reveals distinct transcriptional and post-transcriptional regulatory variation of gene expression and identifies putative alternative splicing variation mediating complex trait variation in pigs
BMC Genomics (2023)
-
Differential adaptive RNA editing signals between insects and plants revealed by a new measurement termed haplotype diversity
Biology Direct (2023)
-
Comprehensive mapping of exon junction complex binding sites reveals universal EJC deposition in Drosophila
BMC Biology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
