
Abstract
Tumour cells become addicted to the expression of initiating oncogenes like Ras, such that loss of oncogene expression in established tumours leads to tumour regression1. HRas, NRas or KRas are mutated to remain in the active GTP-bound oncogenic state in many cancers2. Although Ras activates several proteins to initiate human tumour growth, only PI3K, through activation of protein kinase B (PKB; also known as AKT), must remain activated by oncogenic Ras to maintain this growth3. Here we show that blocking phosphorylation of the AKT substrate, endothelial nitric oxide synthase (eNOS or NOS3), inhibits tumour initiation and maintenance. Moreover, eNOS enhances the nitrosylation and activation of endogenous wild-type Ras proteins, which are required throughout tumorigenesis. We suggest that activation of the PI3K–AKT–eNOS–(wild-type) Ras pathway by oncogenic Ras in cancer cells is required to initiate and maintain tumour growth.
Similar content being viewed by others
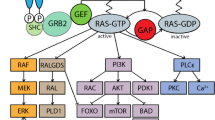
Main
The reduction of Ras oncogene dependence to activation of AKT appears to be a consequence of redundant signalling provided by the established tumour microenvironment. Indeed, activation of AKT fosters tumorigenic growth of otherwise non-tumorigenic cells, provided such cells are mixed with tumour cells to establish the tumour microenvironment3. We exploited this cell-mixing assay to interrogate the signalling pathway downstream of AKT required for tumour maintenance. Although AKT can phosphorylate several proteins4, we focused on BAD, FOXO, IKKα, TSC2 and eNOS, as the consequence of AKT phosphorylation of these proteins is not redundant with the functions of the oncoproteins expressed in cells used in the cell-mixing assay of tumour maintenance5 (Supplementary Fig. 1). Non-tumorigenic PI3K-TtHLacZ cells, derived from normal human kidney cells transformed by oncoproteins T/t-Ags and immortalized by hTERT (hereafter termed TtH cells), and which expressed p110-CAAX (to activate the PI3K-AKT pathway) and LacZ (to demark the cells in the tumour), had Bcl-XL short hairpin RNA (shRNA), eNOS shRNA or, as reported by others6,7,8, dominant-acting FOXO3a-A3 or TSC2SA,TA (mutated at AKT phosphorylation sites), or IKKαK44A (kinase-inactive) proteins expressed to suppress the effects of AKT on these individual pathways (Supplementary Fig. 1). Knockdown of Bcl-XL and eNOS, ectopic expression and nuclear localization of FOXO3a-A3 (ref. 6), ectopic expression of TSC2SA,TA leading to mTOR repression, as assessed by decreased S6k phosphorylation7, and ectopic expression of IKKαK44A leading to repression of NF-κB, as assessed by nuclear exclusion of p658, were validated (Supplementary Fig. 2). These five cell lines were mixed with tumorigenic HRasG12V-transformed TtH cells (termed RasG12V-TtH cells) to establish a tumour microenvironment, injected into mice, and assayed for their contribution to the resultant tumour mass by treating tumours or derived tumour cells with X-gal to stain LacZ+ cells blue. Positive control vector PI3K-TtHLacZ cells extensively populated tumours, whereas negative control vector TtHLacZ cells contributed little to the tumour mass, as evidenced by the prominent or weak blue staining, respectively. Expression of IKKαK44A had little effect; Bcl-XL shRNA, FOXO3a-A3 and TSC2SA,TA had a mild effect; however, eNOS shRNA had the greatest effect on reducing the contribution of PI3K-TtHLacZ (blue) cells in tumours (Fig. 1a).

a, PI3K-TtHLacZ or c, TtHLacZ cells expressing indicated constructs, were mixed with RasG12V-TtH cells, injected into mice, and tumours or re-cultured tumour cells were stained with X-gal (n = 5, mean ± s.e.m.). b, Protein levels of phosphorylated AKT (p-AKT), phopshorylated eNOS (p-eNOS; ect, ectopic; end, endogenous), HA–eNOS (HA) and AKT in PI3K-TtHLacZ cells expressing wild-type or S1177A HA–eNOS treated with DMSO or LY294002.
To test if AKT phosphorylation, not just expression of eNOS, is required for tumour maintenance, AKT was validated to phosphorylate S1177 of eNOS9,10,11. This was evidenced by a loss of S1177 phosphorylation of endogenous eNOS upon pharmacological inhibition of AKT signalling with LY294002, or by mutating S1177 of ectopic eNOS to alanine in PI3K-TtHLacZ cells (Fig. 1b). PI3K-TtHLacZ cells in which eNOS was knocked down were then engineered to express eNOSR in the wild-type or S1177A mutant configuration (Supplementary Fig. 3), and assayed for tumour maintenance by the aforementioned cell-mixing assay. Control PI3K-TtHLacZ cells populated tumours, and this contribution was greatly reduced upon knockdown of eNOS, as evidenced by the reduction in blue staining. This loss was rescued by wild-type, but not S1177A, mutant eNOSR (Fig. 1c). Thus, activation of the PI3K–AKT–eNOS pathway promotes tumour maintenance.
eNOS has been detected in tumour cells12, and catalyses the synthesis of nitric oxide. This can facilitate S-nitrosylation of the thiol group of cysteines in proteins13, such as that of C118 of HRas, which enhances the dissociation of guanine nucleotides, thereby increasing GTP-bound HRas14. Wild-type Ras proteins can be required for activation of the MAPK pathway by oncogenic Ras15 and membrane targeting of RasGAP, which inhibits wild-type but not oncogenic Ras, reverts oncogenic Ras transformation of NIH3T3 cells16. This suggests that wild-type Ras proteins may facilitate oncogenic signalling. Collectively, we speculated that AKT activation of eNOS maintains tumour growth in the absence of oncogenic Ras by activating wild-type Ras through S-nitrosylation of C118. To test this, activated AKT in PI3K-TtH was shown to foster HRas nitrosylation through eNOS. Specifically, most HRas nitrosylation was lost by treatment with the PI3K inhibitor wortmannin (Fig. 2a), by mutating C118 in HRas to serine (a minor change that exchanges the sulphur atom for oxygen but nevertheless blocks nitrosylation14) (Fig. 2b), or by knocking down eNOS (Fig. 2c). Conversly, HRas nitrosylation was elevated upon activation of AKT by p110-CAAX (Fig. 2c). Reduction of HRas nitrosylation by eNOS shRNA also reduced levels of active GTP-bound HRas (Fig. 2c). Because TtH cells express HRas and NRas, but not KRas (not shown), and C118 is conserved among all Ras proteins, we tested and confirmed that activated AKT in PI3K-TtH cells also led to elevated levels of nitrosylated and GTP-bound endogenous NRas, which were reduced upon knockdown of eNOS (Fig. 2c). Thus, AKT activation of eNOS promotes nitrosylation and activation of wild-type Ras proteins. Then, to assess the biological consequence of S-nitrosylation of wild-type HRas in tumour maintenance, we tested whether replacing endogenous wild-type HRas with the nitrosylation-resistant C118S mutant version reduced tumour maintenance. HRas was knocked down by shRNA in PI3K-TtHLacZ cells and complemented with vector encoding an shRNA-resistant HRas (HRasR) in the wild-type or C118S mutant configuration that reduced GTP loading, or, as a control, no transgene (Supplementary Fig. 4). These cell lines, or as control cells expressing either a scramble control sequence or HRas shRNA alone, were mixed with RasG12V-TtH cells, injected into mice, and the resultant tumours assayed for the presence of blue LacZ+ cells as a measure of tumour maintenance. Knockdown of wild-type HRas reduced the ability of PI3K-AKT signalling to foster tumour maintenance, as evidenced by a sixfold reduction of PI3K-TtHLacZ (blue) cells in the tumours. This effect was reversed upon expressing wild-type HRasR, but less so with the C118S mutant version of HRasR (Fig. 2d). Thus, activation of the PI3K-AKT-eNOS pathway promotes tumour maintenance by S-nitrosylation and activation of wild-type Ras.

a–c, Protein levels of S-nitrosylated (nitroso), GTP-bound, total or input HRas or NRas, phosphorylated AKT (p-AKT), or as a loading control tubulin in PI3K-TtHLacZ cells treated with DMSO or wortmannin (a), PI3K-TtHLacZ cells transfected with wild-type or C118S HRas (b), or TtHLacZ cells expressing the indicated combinations of p110-CAAX, vector or eNOS shRNA (c). d, RasG12V-TtH cells were mixed with PI3K-TtHLacZ cells expressing the indicated constructs, injected into mice, and tumours or re-cultured tumour cells were stained with X-gal to visualize PI3K-TtHLacZ cells n = 5, mean ± s.e.m.).
As oncogenic Ras must activate the PI3K-AKT pathway both to initiate and maintain tumour growth3, we tested whether AKT-mediated activation of eNOS was also required for the establishment of tumours. A scramble control or eNOS shRNA was introduced into tumorigenic RasG12V-TtH cells, and knockdown of eNOS complemented by RNAi-resistant eNOS (eNOSR) in the wild-type or S1177A mutant configuration resistant to S1177 phosphorylation (Supplementary Fig. 5). These four cell lines were injected into mice, and tumour growth monitored over time. Scramble control cells rapidly formed tumours, whereas tumour growth was almost abolished upon knockdown of eNOS. This loss of tumour growth was rescued by the wild-type, but not the S1177A version, of eNOSR (Fig. 3a and Supplementary Fig. 6), indicating that S1177 phosphorylation of eNOS is required for tumour initiation and maintenance. These results were validated in a chemical carcinogen-induced spontaneous Ras-driven cancer model. DMBA (7,12-dimethylbenz(a)anthracene) followed by 12-O-tetradecanoylphorbol-13-acetate (TPA) were topically applied to eNOS+/+ and eNOS-/- mice to induce skin papillomas characterized by Ras oncogenic mutations17. The result was an approximate threefold drop in the number of tumours per eNOS-/- mouse (Fig. 3b and Supplementary Fig. 7). Thus, independent models of cancer demonstrate eNOS is required for tumorigenesis.

Representative mice and/or tumours: a, after injection with RasG12V-TtH cells expressing indicated constructs; or b, after treatment with DMBA/TPA to induce skin tumours on mice of indicated genotype (week 20).
To test whether eNOS mediates oncogenic Ras signalling in a cancer associated with the most commonly mutated Ras family member, KRas2, the amount of activated (S1177 phosphorylated) eNOS was first assayed in cancer cell lines and tumour specimens isolated from patients diagnosed with pancreatic cancer. Compared with normal tissue specimens, CFPac-1, MIAPaCa-2 and Capan-1 cells exhibited the highest level of S1177 phosphorylation of eNOS (Fig. 4a). Activated KRas and S1177-phosphorylated eNOS were also elevated in the tumour specimens compared with matched and unmatched normal tissue controls (Fig. 4b and ref. 18), with the caveat that biopsies also contained stromal tissue that could have contributed to detected eNOS phosphorylation.

a, b Protein levels of phosphorylated eNOS (p-eNOS) or as a loading control, tubulin, in pancreatic cancer lines (a), tumour, and normal tissue (b). c–f, Protein levels of GTP-bound or total HRas, NRas or KRas, p-eNOS or tubulin, and excised tumours of CFPac-1 cells expressing eNOS or scramble shRNA (c), CFPac-1 and MIAPaCa-2 cells expressing dox-inducible shRNA +/- dox (d), and CFPac-1 cells expressing HRas (e) or NRas shRNA (f) plus a vector or an RNAi-resistant wild-type or C118S HRas or NRas, or a scramble sequence. g, Proposed signalling.
Next, we tested whether eNOS was required for pancreatic tumour growth. Knockdown of eNOS reduced tumour growth by 50-fold in CFPac-1 cells, and tumour size of MIAPaCa-2 cells by 80% (Fig. 4c, and Supplementary Figs 8 and 9). As a control, knockdown of eNOS in cell lines AsPc-1 and SW1990, which exhibited poor eNOS activation, had no obvious effect on tumour growth (data not shown). Using a more direct assay for tumour maintenance, in vivo doxycycline (dox)-induced shRNA knockdown of eNOS in CFPac-1 and MIAPaCa-2 cells after tumours were established inhibited tumour growth, as evidenced by reduced size and/or gross necrosis of tumours excised at the termination of the experiment (Fig. 4d). In some mice, this eventually led to tumour regression (data not shown). Thus, eNOS is required both to initiate and maintain tumour growth of these human pancreatic cancer cells.
To test whether eNOS promoted tumour growth through nitrosylation of Ras in pancreatic cancer cells, we determined which Ras family members were inactivated by eNOS shRNA in CFPac-1 and MIAPaCa-2 cells. Not surprisingly, GTP-bound KRas was unchanged on knockdown of eNOS (Fig. 4c and Supplementary Fig. 9), as KRas is mutated to remain active in these two cell lines19,20. Consistent with this, oncogenic KRas harbouring the C118S mutation remained tumorigenic (Supplementary Fig. 10), pointing towards wild-type Ras proteins as the target of eNOS signalling. Indeed, GTP-bound endogenous wild-type HRas and NRas were reduced upon shRNA knockdown of eNOS. Moreover, as the wild-type allele of KRas is deleted in MIAPaCa-2 cells20, several oncogenic KRas-positive cell lines20 and tumour tissues21, wild-type HRas and NRas, but not KRas, appear to be the targets of eNOS signalling in pancreatic cancer cells (Fig. 4c and Supplementary Fig. 9).
To test if activation of HRas or NRas by eNOS is required for pancreatic tumour growth, HRas or NRas were knocked down by shRNA in CFPac-1 and/or MIAPaCa-2 cells and complemented with an HRas or NRas that was engineered to be resistant to RNAi (HRasR, NRasR) in the wild-type or C118S mutant configuration. Resultant cells were then assayed for tumour growth in mice. Positive control, scramble-treated CFPac-1 and/or MIAPaCa-2 cells readily formed tumours in mice, whereas this growth was reduced when endogenous HRas, and to a lesser degree NRas, was knocked down. This loss of tumour growth was rescued by expressing the appropriate wild-type HRas or NRas, but not the C118S nitrosylation mutants (Fig. 4e, f and Supplementary Figs 11–13). Similar results were found when the cells were assayed for transformed growth in vitro, suggestive of a tumour-cell autonomous defect when wild-type Ras proteins cannot be nitrosylated (Supplementary Fig. 14). Thus, oncogenic KRas-driven pancreatic cancer tumour growth was mediated by eNOS nitrosylation of endogenous wild-type HRas and NRas (Fig. 4g).
In summary, we demonstrate that the continual need for PI3K-AKT signalling during initiation and maintenance of oncogenic Ras-driven tumour growth is due, at least in part, to activation of eNOS through phosphorylation of S1177. This in turn leads to S-nitrosylation at C118 and correspondingly activation of the other wild-type Ras family members, perhaps as a means to diversify the Ras signal beyond that of oncogenic Ras (Fig. 4g). In agreement, the wild-type counterpart of oncogenic Ras is not required for tumorigenesis (Supplementary Fig. 15), and is even deleted in some tumours21,22; whereas wild-type HRas and NRas are required for oncogenic KRas-driven tumour growth, and appear to have non-redundant activies23,24,25,26. Effects of eNOS on tumorigenesis have been largely attributed to its activity in endothelial cells12. Our results now suggest a key role for tumour-expressed eNOS in the tumorigenic process. Because eNOS plays multiple roles in tumorigenesis12, and delivery of a peptide fragment of the protein cavtratin, which can inhibit eNOS, displays anti-tumour activity27, we speculate that inhibition of eNOS, perhaps in combination with inhibition of wild-type Ras protein function or processing2, could have therapeutic value in the treatment of oncogenic Ras-driven human cancers such as those of the pancreas.
Methods Summary
TtH and the pancreatic cancer cell lines were stably infected retroviruses encoding the indicated shRNAs, transgenes or no insert as described5, and appropriate expression verified by immunoblot or polymerase chain reaction with reverse transcription (RT–PCR). Detection of GTP-bound or nitrosylated Ras was performed as described in Methods. One or a mixture of two cell lines were injected into the flanks of immunocompromised mice to assay for tumour growth; where indicated, they were excised and assayed for LacZ-positive cells, as described3. Induction of shRNA in vivo by dox3 and DMBA/TPA treatments28 were performed as described.
Online Methods
Plasmids
pBabepuro, neo, bleo and hygro were used as control vectors3. The following complementary DNAs (cDNAs) were subcloned into one of the aforementioned pBabe vectors: HA–IKKαK44A cDNA29, FOXO3a-A3 cDNA30 engineered with an amino (N)-terminal HA tag, Flag–TSC2S393A,T1462A cDNA7 (termed here as TSC2SA,TA), eNOS cDNA engineered with a carboxy (C)-terminal HA tag and to be resistant to shRNA by introducing the three silent mutations G1821→A, T1827→C and G1830→A alone (eNOSR) or in conjunction with the mutation A3519GC→GCC that altered S1177 to A (S1177A eNOSR), and wild-type Flag-epitope tagged HRas or NRas cDNAs engineered to be resistant to shRNA by introducing the silent mutations in the region targeted by RNAi (Flag–HRasR; Flag–NRasR) alone or in conjunction with the mutation T342GT→TCT (C118S Flag–HRasR, C118S Flag–NRasR) that altered C118 to S. Bcl-XL shRNA (5′-AGCGTAGACAAGGAGATGC), eNOS shRNA (5′-AAGAGTTATAAGATCCGCTTC), HRas shRNA (5′-GGCAAGAGTGCGCTGACCATC), NRas shRNA (5′-CAAGAAGAGTACAGTGCCATG) or eNOS scramble control (5′-AAGCGTTAAAAGATCCGCTTC) sequences were cloned into pSUPER-PURO-RETRO (Oligoengine). The plasmid system for dox-inducible shRNA3 was adapted to encode eNOS shRNA.
Cell lines
TtH and the pancreatic cancer cell lines were previously described31. Derived lines were generated by stable infection with the indicated combinations of amphotrophic retroviruses generated from the aforementioned pBabe plasmids, as previously described5.
Cell treatments
Cells were treated with LY294002 (Cell Signaling Technologies) or wortmannin (Sigma) at a final concentration of 20 μM or 10 nM, respectively, for 1 h before analysis.
Immunoblotting
HA–IKKαK44A, FOXO3a-A3–HA, HA–eNOS or variants thereof, Flag–TSC2SA,TA, endogenous Bcl-XL, p70 S6 kinase, T389 phosphorylated p70 S6 kinase, HRas, KRas or NRas, S1177 phosphorylated eNOS (both to detect activated eNOS and assess eNOS expression), S473 phosphorylated AKT, actin, p65 and tubulin were detected by immunoblotting with anti-HA (Roche), anti-Flag (Sigma), anti-Bcl-xL, anti-p70 S6 Kinase, anti-Thr389 Phospho-p70 S6 Kinase, anti-Ser1177 Phospho-eNOS, anti-Ser473 Phospho-AKT (Cell Signaling Technology), anti-HRas, anti-KRas, anti-NRas, anti-actin (Santa Cruz), anti-p65 (Rockland) and anti-tubulin (Sigma) antibodies, respectively.
RT–PCR
eNOS and GAPDH mRNA was RT–PCR amplified with the primers 5′-CAGTGTCCAACATGCTGCTGGAAATTG and 5′-TAAAGGTCTTCTTCCTGGTGATGC, and the primers 5′-ACCACAGTCCATGCCATCAC and 5′-TCCACCACCCTGTTGCTGTA, respectively.
GTP and nitrosylated Ras
GTP-bound or nitrosylated Ras were captured as previously described32,33 and immunoblotted with either an anti-Flag (Sigma) or an anti-HRas, anti-KRas or anti-NRas (Santa Cruz) antibody to detect Flag–H-Ras or endogenous H, N or KRas proteins, respectively.
Soft agar
Soft agar assays were done in triplicate and twice independently as previously described5.
Tumour growth
As previously described3, the tested cell line (tumour initiation) or a mixture of two cell lines (cell mixing assay for tumour maintenance) were injected subcutaneously into four flanks of SCID/Beige mice. For tumour initiation experiments, tumours were removed and photographed when control tumours reached maximum volume. For cell mixing assays, the four tumours were removed when they reached maximum volume, human cells derived from the two tumours by re-culture in selective media (G418), and the two other whole tumours were treated with X-gal to stain LacZ+ cells blue and photographed. CFPac-1 and MIAPaCa-2 cells engineered to contain a dox-inducible eNOS shRNA3 were injected into both flanks of five SCID/Beige mice. Tumours were allowed to reach a diameter of 0.6 cm, after which three mice were provided with doxycycline in their diet and two mice left untreated for 11 days (CFPac-1 cells) or 13 days (MIAPaCa-2 cells), after which tumours were removed and photographed. DMBA/TPA treatments were performed as previously described28 on 15 eNOS+/+ C57BL/6J and 15 eNOS-/- C57BL/6J (B6.129P2-Nos3tm1Unc /J) mice34 (Jackson Laboratory). All animal work was approved by the Duke University Medical Center Institutional Animal Care and Use Committee.
Tumour and normal human specimens
Flash-frozen tissue samples were provided devoid of all identifying information under a Duke University Medical Center approved Institutional Review Board protocol.
References
Giuriato, S. et al. Conditional animal models: a strategy to define when oncogenes will be effective targets to treat cancer. Semin. Cancer Biol. 14, 3–11 (2004)
Downward, J. Targeting RAS signalling pathways in cancer therapy. Nature Rev. Cancer 3, 11–22 (2003)
Lim, K. H. & Counter, C. M. Reduction in the requirement of oncogenic Ras signaling to activation of PI3K/AKT pathway during tumor maintenance. Cancer Cell 8, 381–392 (2005)
Luo, J., Manning, B. D. & Cantley, L. C. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell 4, 257–262 (2003)
O’Hayer, K. M. & Counter, C. M. A genetically defined normal somatic human cell system to study ras oncogenesis in vitro and in vivo. . Methods Enzymol. 407, 637–647 (2006)
Brunet, A. et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868 (1999)
Manning, B. D. et al. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 10, 151–162 (2002)
Regnier, C. H. et al. Identification and characterization of an IκB kinase. Cell 90, 373–383 (1997)
Michell, B. J. et al. The Akt kinase signals directly to endothelial nitric oxide synthase. Curr. Biol. 9, 845–848 (1999)
Fulton, D. et al. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 399, 597–601 (1999)
Dimmeler, S. et al. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399, 601–605 (1999)
Fukumura, D., Kashiwagi, S. & Jain, R. K. The role of nitric oxide in tumour progression. Nature Rev. Cancer 6, 521–534 (2006)
Hess, D. T. et al. Protein S-nitrosylation: purview and parameters. Nature Rev. Mol. Cell Biol. 6, 150–166 (2005)
Lander, H. M. et al. Redox regulation of cell signalling. Nature 381, 380–381 (1996)
Hamilton, M. & Wolfman, A. Ha-ras and N-ras regulate MAPK activity by distinct mechanisms in vivo . Oncogene 16, 1417–1428 (1998)
Huang, D. C., Marshall, C. J. & Hancock, J. F. Plasma membrane-targeted ras GTPase-activating protein is a potent suppressor of p21ras function. Mol. Cell. Biol. 13, 2420–2431 (1993)
Quintanilla, M. et al. Carcinogen-specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nature 322, 78–80 (1986)
Lim, K. H. et al. Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr. Biol. 16, 2385–2394 (2006)
Moore, P. S. et al. Genetic profile of 22 pancreatic carcinoma cell lines. Analysis of K-ras, p53, p16 and DPC4/Smad4. Virchows Arch. 439, 798–802 (2001)
Kita, K. et al. Growth inhibition of human pancreatic cancer cell lines by anti-sense oligonucleotides specific to mutated K-ras genes. Int. J. Cancer 80, 553–558 (1999)
Wan, J. et al. Loss of heterozygosity of Kras2 gene on 12p12–13 in Chinese colon carcinoma patients. World J. Gastroenterol. 12, 1033–1037 (2006)
Li, J. et al. LOH of chromosome 12p correlates with Kras2 mutation in non-small cell lung cancer. Oncogene 22, 1243–1246 (2003)
Parikh, C., Subrahmanyam, R. & Ren, R. Oncogenic NRAS, KRAS, and HRAS exhibit different leukemogenic potentials in mice. Cancer Res. 67, 7139–7146 (2007)
Esteban, L. M. et al. Targeted genomic disruption of H-ras and N-ras, individually or in combination, reveals the dispensability of both loci for mouse growth and development. Mol. Cell. Biol. 21, 1444–1452 (2001)
Johnson, L. et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 11, 2468–2481 (1997)
Fotiadou, P. P. et al. Wild-Type NRas and KRas perform distinct functions during transformation. Mol. Cell. Biol. 27, 6742–6755 (2007)
Gratton, J. P. et al. Selective inhibition of tumor microvascular permeability by cavtratin blocks tumor progression in mice. Cancer Cell 4, 31–39 (2003)
Ancrile, B., Lim, K. H. & Counter, C. M. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 21, 1714–1719 (2007)
Woronicz, J. D. et al. IκB kinase-β: NF-κB activation and complex formation with IκB kinase-α and NIK. Science 278, 866–869 (1997)
Hu, M. C. et al. IκB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell 117, 225–237 (2004)
Lim, K. H. et al. Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell 7, 533–545 (2005)
de Rooij, J. & Bos, J. L. Minimal Ras-binding domain of Raf1 can be used as an activation-specific probe for Ras. Oncogene 14, 623–625 (1997)
Jaffrey, S. R. et al. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nature Cell Biol. 3, 193–197 (2001)
Shesely, E. G. et al. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc. Natl Acad. Sci. USA 93, 13176–13181 (1996)
Acknowledgements
We thank J. S. Stamler for human eNOS, L. C. Cantley for TSC2S393A,T1462A, A. Baldwin for IKKαK44A, K. Walsh for FOXO3a complementary DNAs, A. D. Proia for tissue specimens, X.-F. Wang, T.-P. Yao, A. M. Pendergast, C. J. Der, A. D. Cox and M. A. Hollingsworth for discussions, and C. Ring for technical assistance. This research was supported by the NIH and NCI. C.M.C. is a Leukemia and Lymphoma Society Scholar, D.F.K. is a Leukemia and Lymphoma Society Fellow, and K.-H.L. and B.B.A. are Department of Defense Breast Cancer Research Predoctoral Scholars.
Author Contributions K.-H.L., B.B.A. and D.F.K. performed the experiments. All authors conceived and designed experiments and participated in the writing of the paper.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Figures
The file contains Supplementary Figures 1-15 with Legends. (PDF 7094 kb)
Rights and permissions
About this article
Cite this article
Lim, KH., Ancrile, B., Kashatus, D. et al. Tumour maintenance is mediated by eNOS. Nature 452, 646–649 (2008). https://doi.org/10.1038/nature06778
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature06778
This article is cited by
-
Critical requirement of SOS1 for tumor development and microenvironment modulation in KRASG12D-driven lung adenocarcinoma
Nature Communications (2023)
-
Co-targeting KRAS G12C and EGFR reduces both mutant and wild-type RAS-GTP
npj Precision Oncology (2022)
-
The role of ral signaling and post translational modifications (PTMs) of Ras in cancer
Genome Instability & Disease (2022)
-
Post-translational modification of KRAS: potential targets for cancer therapy
Acta Pharmacologica Sinica (2021)
-
Engineering subtilisin proteases that specifically degrade active RAS
Communications Biology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
