
Abstract
Since the completion of the sequencing of the human genome, scientific focus has shifted from studying genes to analysing the much larger number of proteins encoded by them. Several proteins can be generated from a single gene depending on how the genetic information is read (transcribed) and how the resultant protein is modified following translation (post-translational modification). Genomic and proteomic technologies are already providing useful information about autoimmune disease, and they are likely to lead to important discoveries within the next decade.
Similar content being viewed by others
Main
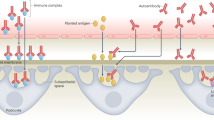
Autoimmune diseases result from three interacting components: genetic, environmental and regulatory1 (Fig. 1). Autoimmunity is caused by a complex interaction of multiple gene products, unlike immunodeficiency diseases, where a single dominant genetic trait is often the main disease determinant2. High-throughput analysis can tell us which genes are turned on or off in different tissues from patients with autoimmune disease or in cells following different stimuli, but analysis of messenger RNA (mRNA) expression alone is insufficient to determine whether the proteins encoded are synthesized. Through recently developed technology we can analyse protein expression and use the information gained to complement the gene-expression findings.

The environment can trigger autoimmunity in genetically predisposed individuals under conditions of immune dysregulation.
These two major investigational approaches, genomic and proteomic, have great potential to shed light on the molecular basis of autoimmune disease in a genetically diverse population. New techniques are likely to markedly accelerate the rate of discovery and characterization of disease-specific genetic and metabolic pathways, and will lead eventually to the development of individualized therapies that take into account markers of disease predisposition and therapeutic response. This review focuses on the main technologies that are being applied to dissect the genome and proteome in autoimmunity. Emphasis is placed on emerging techniques, and the controversial aspects of genomics and proteomics research are discussed.
Genomics and autoimmunity
Traditional methods of differential cloning have been employed successfully to isolate unique genes associated with disease. But these techniques have limited use in the study of multigenic diseases such as autoimmunity. Complementary DNA (cDNA) microarrays, with their ability to determine the expression pattern of thousands of genes simultaneously and to obtain molecular signatures of the state of activity of a cell, are better suited to such studies. It is generally recognized that expression of several genes is coordinated both spatially and temporally and that this coordination changes during the development and progression of disease. Microarray analysis should provide valuable information on disease pathology, progression, response to treatment and overall cellular microenvironments and should also lead to improved, timely diagnosis and novel therapeutic approaches for autoimmune diseases. The genetic datasets obtained, however, are usually highly complex, and the assignment of biological function to the new genes requires other biological methods, such as proteomic analysis, before genetic products can be placed in functional classes or attributed precise roles in cellular pathways.
With the aid of high-throughput sequencing and associated bioinformatic resources, complete sequences of entire genomes, including the human genome (for example, http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=Genome or http://www.ensembl.org), are now available in an organized form within public databases. Concomitant with the development of these new resources, several methods of comparative gene expression have been created to take advantage of the newly available genomic information. Classic quantitative approaches such as northern blotting and RNase protection assays, for which a priori sequence information is required, have been essential tools. The recent development of investigational tools such as differential display, serial analysis of gene expression (SAGE)3 and DNA microarrays4 will enable the investigation of differences in gene-expression patterns that will increase our understanding of autoimmune disease pathogenesis and the construction of disease-specific ‘molecular fingerprinting’ models. When coupled to functional proteomics, these methods gain a second necessary biological dimension as the consequences of altered gene expression can be closely evaluated.
cDNA microarray technology
Microarrays come in two basic varieties: arrays where longer DNA fragments are printed onto a solid support, and arrays where short oligonucleotides are synthesized in situ. The basic concept of data generation is the same: mRNA is reverse-transcribed into cDNA, labelled with a fluorescent dye and hybridized to the array. After washing away any unbound sample, the array is scanned. The fluorescent intensities at a specific spot representing an individual gene directly correlate with the abundance of this gene in the sample. Several methods and software tools have been developed to handle the large volumes of data generated in microarray experiments5. At the simplest level, two samples are compared for differentially expressed genes by calculating the ratios between their fluorescent signals. SAM (significance analysis of microarrays) and other statistical programs allow sophisticated comparison of samples, including assignment of statistical significances to observed differences in gene expression6. Another technique is hierarchical clustering, which uses standard mathematical algorithms to cluster genes with a similar expression pattern across all samples (for example, a time-course experiment) into a dendrogram, where increasing distance between branches reflects increasing dissimilarity of gene-expression patterns7. This method was used to analyse the response of human fibroblasts (cells thought to play a critical role in autoimmune diseases such as scleroderma8) to serum. The most striking finding was the coordinated regulation of expression of genes whose products act at different steps in a common process, such as cell-cycle coordination and proliferation. This suggests that unknown genes within these clusters have a related function. These interrelated gene products, acting together, are called an ‘interactome’ (see below).
Drawbacks of cDNA microarray technology
Unfortunately, cDNA microarray analysis is in its infancy for the study of autoimmune diseases. Although many reports have described the use of commercial microarrays, all of the current studies are plagued with incomplete and potentially misleading data sets. Despite this, several important observations have been reported. Many of the difficulties stem from the use, in current commercial microarrays, of small oligonucleotides that generally correspond to a single exon of a ‘gene’. Thus investigators are generally looking at ‘the ends of genes’. Yet the human and mouse genomes are composed of many segments of coding sequence (exons), interspersed with non-coding segments (introns). A recent genome-wide study of human splicing events demonstrated that at least 74% of human multi-exon genes are alternatively spliced9.
Another problem is that alternative splicing varies depending on the circumstance. For example, under conditions of inflammatory stress, unique isoforms (splice variants) are the rule10. Non-canonical alternative splicing may be an important mechanism for the generation of epitopes to which the immune system is not tolerant, which may lead to autoimmune responses. Current microarray platforms for mammalian gene expression do not allow the identification of splice variants; analyses of many datasets may classify a ‘gene’ as upregulated when it may in fact be a splice variant, expressing the exon defined by the oligonucleotide, but having actions that may not reflect the ‘gene’ that was intended. For example, microarray analysis may indicate that the expression of the gene encoding Otubain-1 is upregulated, as all of its potential 13 variants, which have different (and some opposing) functions, share the same exon defined by the microarray probe11. To overcome this potential for misleading data interpretation, several groups have developed exon-specific arrays and arrays composed of whole-nucleotide sequences to obtain true representation of alternative splicing9,12.
Successful adaptation of cDNA microarray analysis to disease
Global patterns of gene expression can be monitored during disease progression and after clinical intervention. cDNA microarray technology has been successfully used in the clinical setting to study disease biology, especially cancer and autoimmune diseases13,14,15,16,17,18,19,20,21,22. Golub and colleagues reported that mRNA-transcript profiles for leukaemia cells could be divided into acute myeloid and acute lymphoblastic subtypes without prior knowledge21. Alizadeh and colleagues identified distinct signature profiles in diffuse large B-cell lymphomas that correlated with the disease profile22. The Steinman laboratory has used microarray technology for large-scale analysis of mRNA transcripts from complex tissues including human brain specimens from patients with multiple sclerosis13. Other studies using gene microarrays and large-scale robotic sequencing of libraries to study brains of patients with multiple sclerosis or Huntington's disease have also been reported13,14,15. Smaller cDNA arrays have been used in the analysis of gene expression in peripheral blood mononuclear cells of patients with spondyloarthropathy and systemic lupus erythematosus (SLE)16,17. In some cases, important information on the expression of particular clusters of genes, such as anti-apoptotic genes in B-cell lymphomas22, chemokine receptor CXCR4 in arthritis18 and tumour-necrosis factor (TNF)/death receptor family members and interferon-related gene expression in SLE17, has been discovered and has opened up new avenues for therapeutic targets.
Cell lines with a pattern of gene expression observed in autoimmune disease can be screened to select potential drug candidates that can restore the ‘normal’ pattern of expression. Similarly, transgenic animal models expressing or lacking selected genes can be analysed following therapy for modifications to the overall pattern of gene expression that closely resembles that of the affected subject. Gene-expression patterns seen in autoimmune diseases are also reflected in the non-diseased first-degree relatives of the disease probands19.
Defining the interactome
Following the identification of gene-expression patterns by microarray analysis, precise protein–protein interactions need to be worked out to define the interactome. Scientists working on yeast23 and Caenorhabditis elegans24 have pooled their data to generate functional maps of the interactome. But, at present, many data from mammalian studies of protein–protein interaction are not publicly available, and there has been little attempt to share such data. Until this occurs, we will generally depend on the current ‘guilt by association’ model of gene interactions in autoimmune diseases25. This model assumes that proteins with correlated expression levels in microarray analyses26 performed under the same series of conditions are functionally linked. True representation of the interactome will require the analysis of functional links between gene clusters using mating-based yeast two-hybrid assays23,24. This system identifies gene products that interact biochemically at the level of the expressed protein product. By choosing protein domains using available information on amino-acid sequence or secondary structure (such as localization signals, transmembrane regions and domain composition), this technique can be used to study intracellular, transmembrane and secreted protein interactions. Briefly, gene (open reading frame) clones from a cluster of co-expressed genes (obtained from collections such as the I.M.A.G.E consortium (http://image.LLNL.gov) or by polymerase chain reaction) can be fused in-frame with the DNA-binding domain of GAL4, which represents the bait, or to the activation domain of GAL4, which represents the prey. This is carried out in a haploid yeast strain. The bait and prey pools are systematically mated and the transformants selected for the activation of reporter genes. Positive interactions are catalogued, and the resulting binary data can be used in the construction of an interactome. The interactome is then screened against the public literature, where a function may have been noted for one or more members of the network.
As mentioned above, this methodology has been used successfully in yeast and C. elegans23,27,28, as well as in bacteria. Although these organisms have considerably smaller genomes, it should be possible to use this approach to identify functional interactomes in mammalian tissues in autoimmune disease. Interaction mapping of the whole human or mouse genome is not needed; instead, a subset of disease-related genes in relevant tissues can be analysed. For example, inhibition of TNF is beneficial in many autoimmune diseases (see review by Feldmann and Steinman in this issue, page 612), but what is the mechanism for this TNF inhibition? What is the TNF interactome? Does TNF play a direct role in the final common pathway of multiple autoimmune disease effects, or does it act further upstream, disrupting several different pro-inflammatory pathways, each relevant in different therapeutic uses of TNF inhibition? It is only through identification of gene-expression patterns linked to disease pathophysiology, followed by validation of candidate genes through proteomic approaches, that we will realize the success of these new technologies.
Unbiased proteomic technologies
Two major proteomes are the focus of most efforts to understand autoimmune diseases: the serum proteome and the cellular proteome. There are two approaches to investigating each proteome, unbiased and biased. Unbiased technologies attempt to separate and quantify every protein of the expressed proteome (Table 1). This is based on the hypothesis that proteins that are differentially expressed or modified in samples from patients but not in those from healthy controls are likely to be involved in the autoimmune process or at least serve as biomarkers that correlate with disease or therapeutic outcome. Thus, in theory, unbiased approaches have the potential to identify any protein that is involved in autoimmunity without the need for prior knowledge of the protein or its function. In reality, not all proteins are amenable to this kind of analysis because only a fraction of the proteome can be analysed accurately and reliably.
The two most commonly used unbiased technologies are two-dimensional (2D) gel electrophoresis and mass spectrometry (Table 1; ref. 20). With both of these methods, comparisons are made of samples from patients and healthy controls. The main limitations of 2D gel electrophoresis are its low throughput and poor sensitivity, with some estimates suggesting that less than 50% of expressed proteins in cells are amenable to such analysis29. The use of mass spectrometry is plagued by similar, and other, limitations that are beyond the scope of this discussion. An excellent review on this subject has recently been published30.
Despite these limitations, unbiased methods show promise as tools for protein discovery in autoimmunity and have recently led to the identification of candidate markers in the synovial fluid of patients with rheumatoid arthritis31 and the cerebrospinal fluid of patients with multiple sclerosis32. In the case of 2D gel electrophoresis, S100A9, a small calcium-binding protein, was identified as a candidate diagnostic marker for rheumatoid arthritis by comparing the protein profiles of synovial fluid from rheumatoid arthritis and osteoarthritis patients33. In an analogous study using mass spectrometry, C-reactive protein and six members of the S100 protein family (including S100A9) were elevated in the synovial fluid of patients with erosive rheumatoid arthritis compared with that of patients with non-erosive rheumatoid arthritis31. In addition, Stone and colleagues analysed, by mass spectrometry, serum from patients with Wegener's granulomatosis who were enrolled in a trial of the drug etanercept (a TNF inhibitor). They showed that this technique could distinguish between patients in stable clinical remission and those with the active disease34. It is important to note that the proteins or peptides identified using unbiased technologies are simply correlated with disease activity, and that their role in disease causation can only be determined through loss- or gain-of-function experiments. NHLBI's Proteomics Consortium has made a major commitment to these two unbiased approaches, with all but one of the ten centres using one or both of these techniques to understand blood diseases (http://www.nhlbi-proteomics.org/).
Biased proteomic technologies
Once a serum or cellular component of interest is identified using one of several approaches (for example, the unbiased proteomics assays described above, genomic profiling, genetic studies or analysis of knockout animals), methods that enable more focused studies of these ‘known’ components are required — which we term biased approaches (Table 2). The problem with biased approaches is that only a limited subset of proteins to which detection reagents, such as monoclonal antibodies, have been developed can be analysed. Indeed, one of the biggest challenges facing the field of proteomics is the development of high-quality reagents for detecting a large number of proteins.
The main components in the serum proteome that are of particular interest in autoimmunity are autoantibodies and inflammatory mediators, such as cytokines and chemokines. In the cellular proteome, components of interest include T-cell receptors (TCRs), other cell-surface proteins and intracellular signalling proteins. Although this classification scheme is not comprehensive, it provides a useful framework for understanding how different biased technologies can contribute to our understanding of autoimmunity.
The main biased techniques used for large-scale analysis of many proteins are multiplexed western blots35 and protein microarrays36,37. Western blotting is a time-tested technique used in virtually all disciplines of biology. Straightforward protocols have been developed for studying nearly 800 different analytes simultaneously, taking advantage of the fact that proteins migrate at different molecular weights by gel electrophoresis33. Several examples of this technique have now been described for studying autoimmune disease, particularly rheumatoid arthritis33. Lorenz and colleagues compared the proteome in the synovial tissue of patients with rheumatoid arthritis with that of patients with osteoarthritis33. Comparison of the transcriptome and proteome revealed that only 28% of the mRNA and proteins correlated between the patient groups. Moreover, significant differences at the protein level were noted for Stat1, cathepsin D and p47phox, which may be useful targets for therapy.
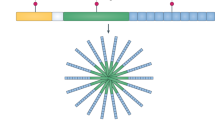
The greatest recent advance in proteomics studies in autoimmunity is the use of protein microarrays. Arrays have been developed specifically for the study of numerous components of both the serum and cellular proteomes, and are described below (Fig. 2).

Discrete components of the autoimmune process from the presentation of MHC–peptide complexes to T cells to the production of autoantibodies by plasma cells can now be analysed in a multiplex fashion using various genomic and proteomic technologies. The numbers in the figure indicate the technologies that have demonstrated potential in the analysis of the corresponding components. (1) Peptide–MHC tetramer arrays. (2) Reverse-phase protein microarrays. (3) Multiparameter flow cytometry for intracellular antigens. (4) cDNA and oligonucleotide microarrays. (5) Antibody microarrays. (6) Bead-based multiplex assays. (7) Autoantigen microarrays. (8) Whole-proteome microarrays. Both mass spectrometry and 2D gel electrophoresis can be used to analyse complex mixtures of proteins and/or peptides.
Antibody profiling using arrays of antigens
Antigen microarrays have been created for a variety of diseases, including infectious diseases (such as HIV; ref. 38), allergies39 and autoimmunity40. Arrays are composed of known antigens, including intact antigenic particles, proteins, lipids, carbohydrates, linear peptides and constrained peptides in which disulphide bonds between cysteine residues provide secondary structure to the peptide40,41. By ‘printing’ all such antigens on the same array, it is possible to gain valuable autoantibody-profiling data with a simple assay. Arrays have been constructed and validated for over a dozen autoimmune diseases, including connective-tissue diseases (such as SLE, scleroderma and myositis), primary biliary cirrhosis, experimental autoimmune encephalomyelitis and multiple sclerosis, rheumatoid arthritis, diabetes42, Crohn's disease, and sclerosing cholangitis40,41,43. Such autoimmune-disease-specific arrays include self-antigens, viral proteins and peptides, and bacterial antigens with complex carbohydrates and recombinant proteins, such as flagellin41.
Arrays of antigens have been used to design and guide development of antigen-specific DNA vaccines for the treatment of multiple sclerosis and HIV infection38. More specifically, arrays of autoantigens or viral antigens are printed and probed using serum derived from animal models of multiple sclerosis (mice with experimental allergic encephalomyelitis) or HIV (macaques with a simian version of HIV), then the antibody response is compared before and after a therapeutic intervention with a DNA vaccine encoding autoantigens or viral proteins, respectively. This line of experimentation has demonstrated that serum antibody epitope spreading is altered as a result of therapy38,43. This technique can be applied to any autoimmune disease in which one or more candidate target antigens have been identified. Large-scale arrays of recombinant proteins can also be produced that may allow the discovery of novel, unidentified autoantigens44. Whether serum autoantibody profiles change in humans in response to therapeutic interventions remains to be studied.
Protein arrays for analysing T-cell receptors
Peptide–major histocompatibility complex (MHC) tetramer arrays have recently been developed and partially validated for the study of antigen-specific T cells45. Individual peptide–MHC tetramer molecules are spotted onto the surface of glass microscope slides at specific positions before being probed with living T cells, which bind to individual tetramer spots and can be quantified and further studied by analysing calcium flux and cytokine secretion. Such assays are required for the detection of immune responses induced by antigen-specific therapies with peptides and proteins and by DNA vaccines (see the review by Feldmann and Steinman in this issue, page 612).
Antibody microarrays
In capture-antibody microarrays, immobilized antibodies trap their specific analytes from a sample solution (for example, serum, synovial fluid, culture supernatant or cell lysates). The bound antigens can then be detected through a direct protein-labelling approach (that is, fluorophores are covalently attached to the antigen) or a sandwich immunoassay approach (that is, a pair of antibodies recognizing two non-overlapping regions of the antigen, where one of them is fluorescently tagged). The intensity of fluorescence corresponds to the concentration of the antigen in the sample. Excellent reviews of this technology have recently been published, and so our discussion here is limited to its relevance to the study of autoimmunity46.
Antibody arrays have been used to analyse intracellular protein levels and even their phosphorylation events, but successful application has largely been limited to the analysis of soluble cytokines and chemokines in culture supernatant and serum samples47. Dysregulation of cytokine signalling is believed to play a crucial role in the initiation and maintenance of autoimmune diseases. For example, several lines of evidence support the notion that interferon-α is central to the development of SLE16,17. Antibody microarrays are particularly suited to revealing unsuspected roles of cytokines in the development of autoimmune diseases. A recent study used antibody microarrays to simultaneously analyse the concentration of 78 cytokines, growth factors and soluble receptors in serum samples derived from patients with Crohn's disease and ulcerative colitis48. Four cytokines were elevated in patients in clinical remission compared with patients with active disease. Among them was transforming growth factor-β (TGF-β), a cytokine that inhibits inflammatory activity and enhances regulatory T-cell functions. Although further studies are required to establish the roles of these cytokines, this is the first successful demonstration of the use of antibody microarrays in the study of autoimmune diseases.
Lysate arrays
One of the most promising techniques for studying blood cells and tissues targeted by the autoimmune response is the reverse-phase protein (RPP) lysate microarray platform49. Lysates prepared from cells are deposited on slides and probed using antibodies of known specificity. Lysates can be prepared from tissue-culture cells, tissue-infiltrating autoreactive lymphocytes, diseased-tissue cells or blood cells and can be stimulated in vitro with agents, such as antigens, cytokines, drugs or antibodies that crosslink cellular receptors50. Antibodies used to probe RPP microarrays include those that recognize housekeeping proteins, inducible factors, signalling proteins, cell-cycle regulatory proteins, apoptosis-related proteins and phosphorylation motifs present in signalling molecules. This approach has been highly successful in characterizing the activation state of tumour cells47 and more recently regulatory T cells50,51. By combining RPP microarrays with laser-capture microdissection (a technique that allows the specific capture and study of individual cells from tissue sections or histological specimens fixed to microscope slides), it should be possible to study rare cells (such as the lymphocytes that infiltrate diseased tissue), dendritic cells and autoimmune target cells, such as glomerular cells in SLE nephritis, neuronal cells in multiple sclerosis or β-cells or islet-infiltrating T cells in insulitis.
Assays based on flow cytometry
Fluorescence-activated cell sorting (FACS) has revolutionized the study of immunology and has recently been adapted for the study of intracellular signalling pathways. In this technique, cell populations are first identified using monoclonal antibodies that are conjugated to spectrally resolvable fluorophores and are specific for cell-surface proteins. Cells are then fixed and permeabilized before staining with antibodies that recognize intracellular proteins, including cytokines, chemokines, structural proteins, apoptosis-related proteins or signalling molecules, such as kinases52. Unlike any of the above technologies, intracellular FACS analysis with multiple fluorochromes allows the characteristics of a single cell to be studied, rather than a mixture of cells. Most recently, this approach has been extended to incorporate the use of phospho-specific antibodies52 and has been validated for characterizing subsets of patients with acute myeloid leukaemia53. Combining this approach with RPP microarrays and multiplexed cytokine assays holds unlimited promise for characterizing autoimmune diseases.
Multiplexed bead-based flow-cytometry assays represent an active area of development. In these assays, differentially identifiable beads are coated with proteins, including antibodies and autoantigens and then identified using a flow cytometer. Such bead-based assays have been adapted for measuring cytokine concentrations in serum or culture supernatants and autoantibodies in serum samples derived from patients with autoimmune disease46. Advances in instrumentation and bead chemistries will probably make this approach very valuable for the study of autoimmunity.
Conclusion and future directions
The expanding use of genomic and proteomic approaches in the analysis of autoimmune disease and therapy has identified numerous areas for improvement and further development. Although DNA microarray studies have led to important advances in the study of autoimmunity, their use is limited owing to the problem of physiologically important splice variants that exist in individual cell populations. In partial response to this problem, several commercial entities are developing new technologies that use exons or single-nucleotide arrays for tiling, allowing additional information on alternative splice variants and polymorphisms. Unfortunately, these new data require additional sophistication in bioinformatics to analyse the increasingly complex data. An additional problem lies in the fact that a large proportion of mRNAs are not translated into proteins, despite being upregulated at the level of transcription.
The proteomics field remains in its infancy and is limited largely by current technology and the dearth of high-quality reagents (specifically monoclonal antibodies) and informatics tools. Unbiased proteomic techniques have largely been disappointing for studying autoimmune diseases, while array and FACS platforms are limited because it is impossible to know which important aspects will be missed owing to the limitations of current arrays. Definition of the interactome is sadly lacking in most mammalian systems. Computing algorithms and two-hybrid systems of interaction need to be generated to allow disparate datasets to be studied simultaneously — such as transcript profiling, protein array and interaction datasets, and FACS. This is no small task. Finally, standards for proteomic work and public datasets need to be developed. Taken together, a multidisciplinary approach to the study of autoimmunity will be required in the coming decade, an approach that combines the skills of biologists, clinicians, engineers and bioinformaticians.
References
Ermann, J. & Fathman, C. Autoimmune diseases: genes, bugs and failed regulation. Nature Immunol. 2, 759–766 (2002).
Buckner, J. & Nepom, G. Genetics of rheumatoid arthritis: is there a scientific explanation for the human leukocyte antigen association? Curr. Opin. Rheumatol. 14, 254–259 (2002).
Ye, S., Usher, D. & Zhang, L. Gene expression profiling of human diseases by serial analysis of gene expression. J. Biomed. Sci. 5, 384–394 (2002).
Moser, K. et al. The use of microarrays to study autoimmunity. J. Invest. Dermatol. Symp. Proc. 1, 18–22 (2004).
Quackenbush, J. Computational analysis of microarray data. Nature Rev. Genet. 2, 418–427 (2001).
Tusher, V., Tibshirani, R. & Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl Acad. Sci. USA 98, 5116–5121 (2001).
Eisen, M., Spellman, P., Brown, P. & Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl Acad. Sci. USA 95, 14863–14868 (1998).
Iyer, V. et al. The transcriptional program in the response of human fibroblasts to serum. Science 283, 83–87 (1999).
Johnson, J. et al. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 302, 2141–2144 (2003).
Ng, B. et al. Increased noncanonical splicing of autoantigen transcripts provides the structural basis for expression of untolerized epitopes. J. Allergy Clin. Immunol. 6, 1463–1470 (2004).
Soares, L. et al. Two isoforms of otubain 1 regulate T cell anergy via GRAIL. Nature Immunol. 1, 45–54 (2004).
Castle, J. et al. Optimization of oligonucleotide arrays and RNA amplification protocols for analysis of transcript structure and alternative splicing. Genome Biol. 10, R66 (2003).
Lock, C. et al. Gene microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nature Med. 8, 500–508 (2002).
Chabas, D. et al. The influence of the proinflammatory cytokine, osteopontin, on autoimmune demyelinating disease. Science 294, 1731–1735 (2001).
Karpuj, M. et al. Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cystamine. Nature Med. 8, 143–149 (2002).
Baechler, E. et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl Acad. Sci. USA 100, 2610–2615 (2003).
Crow, M. & Wohlgemuth, J. Microarray analysis of gene expression in lupus. Arthritis Res. Ther. 6, 279–288 (2003).
Rus, V. et al. Expression of cytokine and chemokine related genes in peripheral blood mononuclear cells from lupus patients by cDNA array. Clin. Immunol. 102, 283–290 (2002).
Aune, T. et al. Co-localization of differentially expressed genes and shared susceptibility loci in human autoimmunity. Genet. Epidemiol. 2, 162–172 (2004).
Robinson, W., Steinman, L. & Utz, P. Proteomics technologies for the study of autoimmune disease. Arthritis Rheum. 46, 885–893 (2002).
Golub, T. et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 286, 531–537 (1999).
Alizadeh, A. et al. distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403, 503–511 (2000).
Ito, T. et al. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl Acad. Sci. USA 98, 4569–4574 (2001).
Li, S. et al. Map of the interactome network of the metazoan C. elegans . Science 303, 540–543 (2004).
Quackenbush, J. Microarrays—guilt by association. Science 302, 240–241 (2003).
Marcotte, E., Pelligrini, M., Thompson, M., Yeates, T. & Eisenberg, D. A combined algorithm for genome-wide prediction of protein function. Nature 402, 83–86 (1999).
Rain, J. et al. The protein-protein interaction map of Helicobacter pylori . Nature 409, 211–215 (2001).
Walhout, A. et al. Protein interaction mapping in C. elegans using proteins involved in vulval development. Science 287, 116–122 (2000).
Gygi, S., Corthals, G., Zhang, Y., Rochon, Y. & Aebersold, R. Evaluation of two-dimensional gel electrophoresis-based proteome analysis technology. Proc. Natl Acad. Sci. USA 97, 9390–9395 (2000).
Coombes, K. R., Morris, J. S., Hu, J., Edmonson, S. R. & Baggerly, K. A. Serum proteomics profiling — a young technology begins to mature. Nature Biotechnol. 23, 291–292
Liao, H. et al. Use of mass spectrometry to identify protein biomarkers of disease severity in the synovial fluid and serum of patients with rheumatoid arthritis. Arthritis Rheum. 50, 3792–3803 (2004).
Dumont, D., Noben, J., Raus, J., Stinissen, P. & Robben, J. Proteomic analysis of cerebrospinal fluid from multiple sclerosis patients. Proteomics 4, 2117–2124 (2004).
Drynda, S. et al. Proteome analysis reveals disease-associated marker proteins to differentiate RA patients from other inflammatory joint diseases with the potential to monitor anti-TNFα therapy. Pathol. Res. Pract. 200, 165–171 (2004).
Stone, J. et al. A serum proteomic approach to gauging the state of remission in Wegener's granulomatosis. Arthritis Rheum. 52, 902–910 (2005).
Lorenz, P., Ruschpler, P., Koczan, D., Stiehl, P. & Thiesen, H. From transcriptome to proteome: differentially expressed proteins identified in synovial tissue of patients suffering from rheumatoid arthritis and osteoarthritis by an initial screen with a panel of 791 antibodies. Proteomics 3, 991–1002 (2003).
Macbeath, G. Protein microarrays and proteomics. Nature Genet. 32, 526–532 (2002).
Wilson, D. & Nock, S. Recent developments in protein microarray technology. Angew. Chem. 42, 494–500 (2003).
de Vegvar, H. et al. Microarray profiling of antibody responses against Simian-Human Immunodeficiency Virus: Postchallenge convergence of reactivities independent of host histocompatibility type and vaccine regimen. J. Virol. 77, 11125–11138 (2003).
Wiltshire, S. et al. Detection of multiple allergen-specific IgEs on microarrays by immunoassay with rolling circle amplification. Clin. Chem. 46, 1990–1993 (2000).
Robinson, W. et al. Autoantigen microarrays for multiplex characterization of autoantibody responses. Nature Med. 8, 295–301 (2002).
Wang, D., Liu, S., Trummer, B., Deng, C. & Wang, A. Carbohydrate microarrays for the recognition of cross-reactive molecular markers of microbes and host cells. Nature Biotechnol. 20, 275–281 (2002).
Quintana, F. et al. Functional immunomics: microarray analysis of IgG autoantibody repertoires predicts the future response of mice to induced diabetes. Proc. Natl Acad. Sci. USA 101, 14615–14621 (2004).
Robinson, W. et al. Protein microarrays guide tolerizing DNA vaccine treatment of autoimmune encephalomyelitis. Nature Biotechnol. 21, 1033–1039 (2003).
Michaud, G. et al. Analysing antibody specificity with whole proteome microarrays. Nature Biotechnol. 21, 1509–1512 (2003).
Soen, Y., Chen, D., Kraft, D., Davis, M. & Brown, P. Detection and characterization of cellular immune responses using peptide-MHC microarrays. PLoS Biol. 1, E65 (2003).
Utz, P. Protein arrays for studying blood cells and their secreted products. Immunol. Rev. 204, 264–282 (2005).
Gilburd, B. et al. Autoantibodies profile in the sera of patients with Sjogren's syndrome: the ANA evaluation—a homogeneous, multiplexed system. Clin. Dev. Immunol. 11, 53–56 (2004).
Kader, H. A. et al. Protein microarray analysis of disease activity in pediatric inflammatory bowel disease demonstrates elevated serum PLGF, IL-7, TFG-β1, and IL-12p40 levels in Crohn's disease and ulcerative colitis patients in remission verus active disease. Am. J. Gastroenterol. 100, 414–423.
Espina, V. et al. Protein microarrays: molecular profiling technologies for clinical specimens. Proteomics 3, 2091–2100 (2003).
Chan, S., Ermann, J., Su, L., Fathman, C. & Utz, P. Protein microarrays for multiplex analysis of signaling pathways. Nature Med. 10, 1390–1396 (2004).
Su, L. et al. Murine CD4+CD25+ regulatory T cells fail to undergo chromatin remodeling across the proximal promotoer region of the IL-2 gene. J. Immunol. 173, 4994–5001 (2004).
Perez, O. & Nolan, G. Simultaneous measurement of multiple active kinase states using polychromatic flow cytometry. Nature Biotechnol. 20, 155–162 (2002).
Irish, J. et al. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell 118, 217–228 (2004).
Petricoin, E. et al. Use of proteomic patterns in serum to identify ovarian cancer. Lancet 359, 572–577 (2002).
Zhu, H. et al. Global analysis of protein activities using proteome chips. Science 293, 2101–2105 (2001).
Joos, T. et al. A microarray enzyme-linked immunosorbent assay for autoimmune diagnostics. Electrophoresis 21, 2641–2650 (2000).
Schweitzer, B. et al. Multiplexed protein profiling on microarrays by rolling circle amplification. Nature Biotechnol. 20, 359–365 (2002).
Liotta, L. et al. Protein microarrays: meeting analytical challenges for clinical applications. Cancer Cell 3, 317–325 (2003).
Acknowledgements
The authors thank members of the Utz and Fathman labs for insights and scientific creativity. CGF is supported by the JDRFI, ADA, UCSF and the NIH. Steven Chan is supported by the Stanford MSTP, Bio-X and the Floren Family Foundation. Luis Soares is supported by the NIH. PJU is the recipient of a Donald E. and Delia B. Baxter Foundation Career Development Award and was supported by the Dana Foundation, the Floren Family Foundation, the Stanford Program in Molecular and Genetic Medicine (PMGM), NIH Grants and NHLBI Proteomics Consortium.
Author information
Authors and Affiliations
Ethics declarations
Competing interests
PJU states the following conflict of interest disclosures, all directly relevent to proteomics technology described in this manuscript: During the past three years, he has served asa consultant to Becton Dickinson Biosciences (San Jose, CA) and Genentech (South San Francisco, CA), is a member of the Scientific Advisory Board of ACLARA Biosciences (Mountain View, CA) ans is a Co-Founder and Consultant at Bayhill Therapeutics (Palo Alto, CA).
Rights and permissions
About this article
Cite this article
Fathman, C., Soares, L., Chan, S. et al. An array of possibilities for the study of autoimmunity. Nature 435, 605–611 (2005). https://doi.org/10.1038/nature03726
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature03726
This article is cited by
-
Secretome analysis of in vitro aged human mesenchymal stem cells reveals IGFBP7 as a putative factor for promoting osteogenesis
Scientific Reports (2018)
-
Design of Chemical Conjugate for Targeted Therapy of Multiple Sclerosis Based of Constant Fragment of Human Antibody Heavy Chain and Peptoid Analog of Autoantigen MOG35-55
Bulletin of Experimental Biology and Medicine (2017)
-
Proteomics-based identification of haptoglobin as a favourable serum biomarker for predicting long-term response to splenectomy in patients with primary immune thrombocytopenia
Journal of Translational Medicine (2012)
-
Gene-expression profiling in rheumatic disease: tools and therapeutic potential
Nature Reviews Rheumatology (2009)
-
Gene expression profiling in streptozotocin-induced diabetic rat liver in response to fungal polysaccharide treatment
Korean Journal of Chemical Engineering (2009)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
