
Abstract
Interactions between microbes and human hosts can range from a benign, even symbiotic collaboration to a competition that may turn fatal — resulting in death of the host, the microbe or both. Despite advances that have been made over the past decades in understanding microbial pathogens, more people worldwide still die every year from infectious disease than from any other cause. This highlights the relevance of continuing to probe the mechanisms used by microorganisms to cause disease, and emphasizes the need for new model systems to advance our understanding of host–pathogen interactions.
Similar content being viewed by others

Main
Although a wide range of microbe–host relationships can ultimately lead to disease, the two most general strategies used by pathogenic microbes may be described in military terms as ‘frontal’ and ‘stealth’ assaults. Pathogenic microorganisms make use of both of these approaches. Typically, frontal assault strategies require that the infecting microbes rapidly replicate, induce disease symptoms that overwhelm the innate defences of the host, and find a new host before engagement of the ‘adaptive’ or ‘acquired’ immune system, in which antigen-specific lymphocytes respond to antigen exposure. Stealth assaults, on the other hand, typically involve a slower infection process in which the microbes subvert the host's innate and adaptive immune systems to set up a chronic or persistent infection (Box 1). The diverse tactics used for both forms of attack have been the subject of intense investigation, which has helped not only to shape our understanding of the invading organisms, but also to define many of the pathways that are important in host resistance and susceptibility, and in normal host physiology. In discussing microbial pathogenicity, it is important to consider that every human is host to myriad microorganisms, and that illness is the exception rather than the rule (Box 2). Nevertheless, microbial pathogens impose a tremendous medical burden, so it is important that we understand their diverse assault strategies. Here we outline a variety of frontal and stealth strategies used by pathogenic microorganisms. In particular, we discuss the less well-known strategies used by chronically infecting bacteria, and outline the mechanisms used by these pathogens to subvert both the innate and the acquired immune systems.
Frontal assault strategies
Many pathogenic microbes adopt an aggressive strategy in which they immediately attack and attempt to overwhelm the host's innate immune system. Although there are many variations to this mode of assault (Table 1), these microbes typically produce toxins or use complex secretion systems to deliver so-called effector proteins, which disrupt the normal function of the host cell.
One example of a pathogen that uses a frontal assault strategy is Vibrio cholerae, a potent epidemic adversary in many developing areas of the world. The organism is typically ingested in contaminated food and water, and produces a powerful toxin that causes a voluminous secretory diarrhoea1. In severely affected patients, the volume of stool purged can be in excess of the patient's own body weight2 and dehydration is the typical cause of death. The ability of V. cholerae to rapidly cause disease before eliciting a productive immune response is shared by many microorganisms, including the most common cause of diarrhoeal disease in infants, the rotavirus family, which is responsible for more than 600,000 deaths a year3.
Cholera toxin is encoded in the genome of a bacteriophage, called cholera-toxin phage, that is integrated into the V. cholerae genome. Thus, the virulence of V. cholerae results from the acquisition, by horizontal genetic transfer, of foreign DNA that encodes a toxin4. The receptor used by the cholera-toxin phage to infect V. cholerae is encoded as part of an additional portion of horizontally acquired DNA known as a pathogenicity island (PAI).
PAIs, found in many types of bacteria, are large chromosomal regions of DNA that are believed to have originated either from bacteriophages or from small, self-replicating bacterial DNA structures called plasmids5. This mechanism, in which acquired DNA has an important role in converting an otherwise harmless species into a pathogen, has been recognized as a common occurrence for many bacteria. It also serves as a microbial example of darwinian natural selection, in which the presence of pathogenic species is partially due to selective pressure applied by the host on the infecting microbe. For example, it is conceivable that before acquisition of the cholera toxin, the ancestral form of V. cholerae would colonize and reside in the small intestine but would cause few clinical symptoms. Eventually, this extended stay would result in engagement of the adaptive immune system. Because V. cholerae has few, if any, mechanisms to subvert the adaptive branch of the human immune system, most of the bacteria would be killed and not disseminated back to the environment. So although it is possible that the human was a ‘dead end’ for the ancestral form of V. cholerae, infection with the cholera-toxin phage produced a strain that could proliferate in the human host and be shed in great numbers back to the aquatic environment before productive engagement of the adaptive immune response. As a result, this pathogen has flourished. This illustrates how host diligence in the form of innate and adaptive immunity imposes selective pressures that shape pathogenic lifestyle, and helps to explain why closely related pathogens have such different lifestyles and cause such diverse diseases.
Many bacterial pathogens have been shown to cause disease by delivering proteins directly into the host cell that they are infecting. This is done using specialized type III/IV secretion systems that are typically acquired as part of a PAI. This island encodes components that assemble into a ‘molecular syringe’, which can inject effector molecules into the host cell6,7. For example, the genus Yersinia contains three pathogenic species that use type III secretion to subvert the host immune response and cause diseases ranging from gastrointestinal disorders to bubonic plague. When it enters the host, Yersinia encounters macrophages and other specialized phagocytic cells, which serve as the first line of immunological defence. Interaction with these host cells causes Yersinia to produce and deliver proteins called Yersinia outer-membrane proteins (Yops), which prevent phagocytosis, disrupt normal signal transduction pathways and initiate the ultimate apoptotic demise of the phagocytic cells6. Unlike V. cholerae, which delivers its potent toxin and is then rapidly shed from the body in the diarrhoeal output, thereby avoiding a strong inflammatory response, Yersinia prefers a ‘stand and fight’ approach in which it faces and overcomes the host's innate and adaptive immune systems.
Stealth assault strategies
Our understanding of the strategies used by pathogens to set up persistent infections remains fairly limited compared with our knowledge of the frontal assault strategies discussed above. It is important to point out that a high proportion of persistent infections are caused by viral and parasitic pathogens8,9,10,11. Nevertheless, chronically infecting bacteria present a significant challenge in terms of their ability to cause disease. The remainder of this review will focus primarily on microbes that use stealth assault strategies to cause chronic infections, and on recent findings that may further our understanding of the complex interplay between host and pathogen.
Helicobacter pylori
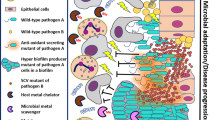
Infecting over 50% of the world's population, Helicobacter pylori has been recognized as something of a bacterial role model for its ability to colonize and persist for the lifetime of the host, even when faced with a robust host immune system. The bacterium exclusively infects human and non-human primates, and persists in the harsh environment of the stomach. About 20% of those infected with H. pylori will ultimately suffer diseases ranging from gastritis and ulcer disease to gastric cancer12. This microbe, which uses a variety of strategies and gene products to subvert the innate and adaptive immune systems of the host (Fig. 1), therefore represents a tremendous medical challenge.

The bacterium overcomes the innate immune system by neutralizing gastric acid (a) and by having lipopolysaccharide and flagella with low immunostimulatory recognition by Toll-like receptors (b). The adaptive immune system is also affected: B-cell (c) and T-cell (d) proliferation are blocked by the activity of CagA (a product of the cytotoxin-associated gene (cag) pathogenicity island) and VacA (vacuolating cytotoxin), respectively. In addition, the bacterium can escape exposure to host immune defences by surviving intracellularly (e), and shows a high frequency of genetic rearrangement (f) that appears to be essential for persistent colonization.
Gastric acidity is a major antimicrobial defence, and the stomach serves as a death chamber for most of the millions of bacteria that are ingested daily. However, H. pylori not only survives the low pH, but thrives in the human stomach. The bacterium has a number of strategies to overcome acid stress13. Foremost among these approaches is production of the bacterial enzyme urease, which catalyses the hydrolysis of urea to carbon dioxide and ammonia, and helps to maintain a protonmotive force, essential for bacterial metabolism and survival, across the H. pylori inner membrane14,15. The production of the basic ammonia molecule helps to buffer the bacterial cytoplasm and the microenvironment as it is secreted from the bacterial cell.
Another important part of the human innate immune system involves Toll-like receptors (TLRs). Bacterial lipopolysaccharide (LPS), a component of the the bacterial cell wall, is typically recognized by TLR4 and this triggers a robust proinflammatory response. However, recent work has shown that primary stomach epithelial cells and gastric epithelial cell lines do not react to the LPS from H. pylori16. In addition, it has been shown that recognition of H. pylori by TLR5 is markedly different from that observed with other Gram-negative pathogens, as H. pylori flagellins do not signal through TLR5 to stimulate an innate immune response17,18. Despite the lack of immunostimulation by these traditional pathways, H. pylori is reported to cause a strong inflammatory response in vivo through nuclear factor (NF)-κB activation. It has been suggested that this is required for establishment of a chronic infection19. So although the bacterium bypasses the innate TLR system, it can engage alternative inflammatory mediators.
H. pylori not only subverts the innate immune system, but also modulates the adaptive immune system by blocking the antigen-dependent proliferation of T cells. This is accomplished partially by delivery of vacuolating cytotoxin, VacA20, which blocks T-cell receptor signalling events that normally lead to the production of cytokines, important mediators of the immune response.
A role in subverting the other branch of the adaptive immune response has been indicated by the finding that ectopic expression in B cells of the CagA protein — a product of the cytotoxin-associated gene (cag) PAI of H. pylori — inhibits B-cell proliferation by suppressing the JAK–STAT signalling pathway21. This study also suggested that CagA represses B-cell apoptosis, which may contribute to the formation of H. pylori-induced mucosa-associated lymphoid tissue (MALT) lymphoma. In this case, host immune defences that are designed to kill the invading microbe are subverted in such a way that they harm the host and not the bacterium.
As well as actively suppressing the immune response and the ability of the host to clear the infection, recent evidence suggests that H. pylori can hide from classic host defences by becoming intracellular. Although H. pylori is believed to remain primarily extracellular during infection, it was noted by early investigators examining gastric biopsies that a subpopulation of the bacteria could be found within cells22,23. The relevance of this observation was questioned for many years, but recent studies have shown that H. pylori can invade cultured eukaryotic cells and reside within multivesicular bodies that promote bacterial survival, motility and replication, and ultimately serve as a reservoir for re-establishment of infection (the intracellular bacteria egress from the vesicles and reseed the extracellular milieu)24. The relevance of these in vitro findings is supported by recent work showing that the bacterium could be detected inside precancerous and cancerous epithelial cells from H. pylori-positive patients25. In addition, the relative level of expression of several known bacterial virulence factors was considerably higher in the advanced-stage cancerous cells, suggesting that expression of virulence factors by these intracellular bacteria may play a part in disease progression. The ability of H. pylori to survive intracellularly and to escape many host immune defences is not unique. It is used by many microbial pathogens, including Mycobacterium tuberculosis, Listeria monocytogenes and Shigella flexneri, the causative agents of tuberculosis, listeriosis and shigellosis, respectively.
A final mechanism that may be crucial for H. pylori to escape the host immune response involves its ability to undergo genetic rearrangement that either eliminates particular immunostimulatory gene products or causes variation in potentially immunostimulatory molecules. Genomotyping of clonal isolates using an H. pylori microarray from a single human host suggests that as many as 3% of the loci show significant genetic variation26. Although the implications of this finding are not fully understood, a study showing that strains of H. pylori that have deleted all or portions of the PAI that delivers CagA to the host cell are more likely to colonize various animal models suggests that the ability of the bacterium to modulate gene products that affect the robustness of the immune response is significant27.
Although CagA is a virulence factor for H. pylori, the presence of CagA is not sufficient to predict the disease outcome in H. pylori infection. It is known that individuals infected with CagA that can be phosphorylated have more severe disease than patients infected with a non-phosphorylateable CagA, but which of these forms of CagA was the progenitor in the evolution of H. pylori is unclear. A functional PAI is found in most human clinical isolates, but there is variation in the cagA coding region in the critical EPIYA domains that are phosphorylated by host tyrosine kinases28.
These epidemiological results are supported by the recent finding that intragenomic recombination in the cagA coding sequence results in the production of a non-phosphorylateable form of CagA that does not induce morphological changes associated with pathogenicity in host cells29. The importance of this strategy of genetic rearrangement within the host may explain the recent identification of the RuvC protein, a Holliday junction resolvase involved in recombination, as a crucial factor in bacterial persistence30.
Many other pathogens use genetic rearrangement as a means of immune escape. Included among these are Neisseria gonorrhoeae and HIV, which are responsible for gonorrhoea and AIDS, respectively.
Salmonella enterica serovar Typhi
Many Salmonella serovars, like Salmonella typhi, the causative agent of typhoid fever, are adapted exclusively to human hosts and are endemic in areas of the world with poor sanitation and inadequate sewage-treatment facilities. Typhoid fever remains an important cause of human morbidity and mortality, and is becoming increasingly difficult to treat owing to the emergence of antibiotic-resistant strains31. Between 1% and 6% of those infected with S. typhi become chronic carriers of the bacterium and, although not necessarily suffering any symptoms themselves, may serve as a reservoir for subsequent infections during periods of faecal and urinal shedding32 — think Typhoid Mary (see Box 3). Sites of persistence within the body have been shown to include the bone marrow and the gall bladder33,34, but little is understood about the adaptive mechanisms used by the bacteria during the course of establishing a persistent infection. Recent work using a novel model of bacterial persistence with S. typhimurium has begun to shed light on Salmonella persistence strategies35 and should be amenable to genetic analysis, both to dissect the role of individual bacterial factors in persistence and to explain the failure of the host immune system to eradicate the infection.
Adaptation of Salmonella serovars to the host and the capacity of adapted microbes to persist are essential survival features of most, if not all, Salmonella serovars. Although S. typhimurium is well known as a cause of food-borne gastroenteritis in humans, this contributes little to the survival of the species because it rarely establishes a carrier state in humans and is associated only occasionally with intrafamilial spread. Mice infected with S. typhimurium have been used as a model to study acute systemic disease, as the bacterium causes a typhoid-like disease in some mouse strains. Differences in mouse susceptibility to S. typhimurium have been linked to the particular allele of the Nramp1 gene being expressed in cells of the monocyte/macrophage lineage36. Nramp1 is involved in controlling exponential growth of the bacteria in the reticuloendothelial system during the early stages of infection. In a recent study, mice expressing the wild-type Nramp1 allele did not die after oral inoculation with S. typhimurium, but became uniformly persistently infected in a manner similar to that observed in chronic typhoid carriers35. In these animals, Salmonella was found to persist predominately in the mesenteric lymph nodes and/or spleen even in the face of a robust antibody response. Closer inspection showed that the bacteria were actually intracellular, having colonized macrophages. Most of the infected macrophages only contained between one and three bacteria — fewer than would be expected on the basis of studies in cultured macrophages, in which the SPI-2 pathogenicity island of Salmonella (a genetic element responsible for causing disease) is thought to permit intracellular replication. Whether SPI-2 is actually expressed in persistent infection of macrophages is not certain. However, reactivation of intracellular Salmonella and systemic spread could be accomplished by administering interferon-γ (IFN-γ)-neutralizing antibodies, suggesting that the host cytokine IFN-γ is important for suppression of Salmonella replication and disease35. IFN-γ is also important for the maintenance of latent infections of Mycobacterium tuberculosis and Chlamydia trachomatis. Using this persistence model and recent technological advances, it should be possible to identify bacterial factors that are important for persistence. This could be accomplished using microarrays and global screening strategies of libraries of defined Salmonella mutants37.
Bartonella spp.
The Bartonella genus is increasingly recognized as a significant human pathogen. Bartonella spp. cause numerous diseases, including Oroya fever, verruga peruana, trench fever, bacillary angiomatosis, endocarditis and cat-scratch disease38. Bartonella infections are disseminated from their natural reservoir (cats, rats, humans, deer and other mammals) by arthropods such as fleas and ticks. Carrier rates in the reservoir population are quite high: as many as 41% of domestic cats have been shown to be infected with B. henselae39. Bartonellae are fairly unique among bacterial pathogens in terms of their ability to sustain prolonged periods of parasitism within red blood cells. They have been shown to invade, multiply within and persist for the lifetime of the infected host cell40, and to reach titres of 104 per ml of blood in infected humans41. How this many Gram-negative organisms can persist within the bloodstream without inducing a classic septic shock response remains to be determined, but the finding suggests that, as with H. pylori, the LPS of Bartonella may have reduced immunostimulatory properties. Also, as for Salmonella, the ability to survive intracellularly helps Bartonella to escape the host immune response. Antibodies have no effect on Bartonella within red blood cells, but may contribute to preventing new waves of bacterial invasion42. It has also been suggested that intracellular Bartonella may affect the development of an adaptive immune response in a manner similar to that seen with malaria-infected red blood cells43. Malaria prevents the maturation of dendritic cells, which are essential for the development of a normal adaptive immune response44.
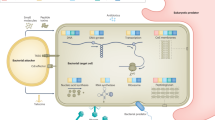
Genetic studies and development of appropriate animal models of haemotropic infection by Bartonella have begun to shed light on some of the bacterial factors that are required for intraerythrocytic infection (Fig. 2). One important virulence factor in Bartonella is the type IV secretion system. It was recently shown that mutation of either virB4 or virD4 genes in B. tribocorum prevents the bacterium from establishing intraerythrocytic bacteraemia in a rat model of infection45 — although whether this failure is due to changes in endothelial cell interaction or the inability to deliver a necessary effector protein remains to be determined. It is notable that another family of zoonotically acquired human pathogens, Brucella, which causes undulant fever in humans, also requires a type IV secretion system to establish infection46.

a, Bartonella colonizes an unknown niche within the body and seeds bacteria that are able to infect and survive inside red blood cells. b, Host antibodies have no effect on intraerythrocytic bacteria but prevent new waves of erythrocytic parasitism. c, d, Intracellular bacteria have been proposed to affect dendritic cell maturation through an unknown mechanism (c), and have been shown to inhibit cytokine production and T-cell proliferation (d) by inducing the expression of interleukin-10 (IL-10). e, A type IV secretion system is essential for establishing persistent intraerythrocytic infection.
In addition to its ability to invade and persist within red blood cells, Bartonella apparently manipulates the host immune system through modification of host-cell cytokine production. It was shown that homeless people who were chronically and asymptomatically infected with B. quintana showed decreased levels of circulating markers of leukocyte activation and decreased cytokine production by mononuclear cells. This was accompanied by an increase in the secretion of interleukin-10 (IL-10)43, which depresses the expression of other cytokines and the release of soluble inflammatory mediators. Activation of T cells in the presence of IL-10 has been shown to result in non-responsiveness of these cells and attenuation of downstream signalling events47. Thus Bartonella-induced expression of IL-10 may directly facilitate bacterial persistence. This increasingly recognized persistence strategy has been observed for Leishmania major, Coxiella burnetii, Yersinia pseudotuberculosis and Onchocerca volvulus, the causative agents of leishmaniasis, Q fever, gastroenteritis and river blindness, respectively. Like Bartonella, these pathogens not only circumvent normal host defences, but also directly manipulate them.
Fighting back
Years of research have helped to define our current understanding of pathogens and host–pathogen interactions. But of the more than 1,400 known bacterial, viral and parasitic pathogens that infect humans, we have managed to completely eradicate only smallpox48. Although the spread of several pathogens has been greatly diminished over the past 30 years, a further 37 human pathogens have been discovered and an estimated 12% of the known pathogens have been recognized as emerging or re-emerging49. About 200 different bacterial species are known to cause human disease48, and these microbes use multiple and diverse strategies to overcome the host immune system. With the continuing rise in infectious-disease-related morbidity and mortality, future research should be directed towards gaining a greater understanding of the host–pathogen interface and strategies used by microbes for modulation of the host immune system. To accomplish this, we must focus on clarifying not only virulence factors per se, but also specific mechanisms that allow microbes to persist within the host environment. Gaining a thorough knowledge of chronically infecting microbes and learning how to control them is perhaps the greatest challenge. It is our belief that this should be made a major research focus and funding priority. We must strive to develop and apply novel technological approaches and new model systems to the elucidation of persistence strategies if we ever hope to find ways to subvert these stealthy pathogens.
References
Kaper, J. B., Fasano, A. & Trucksis, M. in Vibrio cholerae and Cholera: Molecular to Global Perspectives (eds Wachsmuth, I. K., Blake, P. A. & Olsvik, Ø.) 145–176 (ASM Press, Washington DC, 1994).
Phillips, R. A. Water and electrolyte losses in cholera. Fed. Proc. 23, 705–718 (1964).
Wilhelmi, I., Roman, E. & Sanchez-Fauquier, A. Viruses causing gastroenteritis. Clin. Microbiol. Infect. 9, 247–262 (2003).
Waldor, M. K. & Mekalanos, J. J. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 272, 1910–1914 (1996).
Schmidt, H. & Hensel, M. Pathogenicity islands in bacterial pathogenesis. Clin. Microbiol. Rev. 17, 14–56 (2004).
Cornelis, G. R. The Yersinia Ysc–Yop ‘type III’ weaponry. Nature Rev. Mol. Cell Biol. 3, 742–752 (2002).
Christie, P. J. & Vogel, J. P. Bacterial type IV secretion: conjugation systems adapted to deliver effector molecules to host cells. Trends Microbiol. 8, 354–360 (2000).
Brake, D. A. Parasites and immune responses: memory illusion? DNA Cell Biol. 22, 405–419 (2003).
Crowe, S., Zhu, T. & Muller, W. A. The contribution of monocyte infection and trafficking to viral persistence, and maintenance of the viral reservoir in HIV infection. J. Leukoc. Biol. 74, 635–641 (2003).
Jarvis, M. A. & Nelson, J. A. Human cytomegalovirus persistence and latency in endothelial cells and macrophages. Curr. Opin. Microbiol. 5, 403–407 (2002).
Johnson, W. E. & Desrosiers, R. C. Viral persistance: HIV's strategies of immune system evasion. Annu. Rev. Med. 53, 499–518 (2002).
Ernst, P. B. & Gold, B. D. The disease spectrum of Helicobacter pylori: the immunopathogenesis of gastroduodenal ulcer and gastric cancer. Annu. Rev. Microbiol. 54, 615–640 (2000).
Valenzuela, M., Cerda, O. & Toledo, H. Overview on chemotaxis and acid resistance in Helicobacter pylori. Biol. Res. 36, 429–436 (2003).
Weeks, D. L., Eskandari, S., Scott, D. R. & Sachs, G. A H+-gated urea channel: the link between Helicobacter pylori urease and gastric colonization. Science 287, 482–485 (2000).
Mobley, H. L., Island, M. D. & Hausinger, R. P. Molecular biology of microbial ureases. Microbiol. Rev. 59, 451–480 (1995).
Smith, M. F. Jr et al. Toll-like receptor (TLR) 2 and TLR5, but not TLR4, are required for Helicobacter pylori-induced NF-κB activation and chemokine expression by epithelial cells. J. Biol. Chem. 278, 32552–32560 (2003).
Lee, S. K. et al. Helicobacter pylori flagellins have very low intrinsic activity to stimulate human gastric epithelial cells via TLR5. Microbes Infect. 5, 1345–1356 (2003).
Gewirtz, A. T. et al. Helicobacter pylori flagellin evades Toll-like receptor 5-mediated innate immunity. J. Infect. Dis. 189, 1914–1920 (2004).
Rhen, M., Eriksson, S., Clements, M., Bergstrom, S. & Normark, S. J. The basis of persistent bacterial infections. Trends Microbiol. 11, 80–86 (2003).
Gebert, B., Fischer, W., Weiss, E., Hoffmann, R. & Haas, R. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science 301, 1099–1102 (2003).
Umehara, S., Higashi, H., Ohnishi, N., Asaka, M. & Hatakeyama, M. Effects of Helicobacter pylori CagA protein on the growth and survival of B lymphocytes, the origin of MALT lymphoma. Oncogene 22, 8337–8842 (2003).
Bode, G., Malfertheiner, P. & Ditschuneit, H. Pathogenetic implications of ultrastructural findings in Campylobacter pylori related gastroduodenal disease. Scand. J. Gastroenterol. Suppl. 142, 25–39 (1988).
Foliguet, B., Vicari, F., Guedenet, J. C., De Korwin, J. D. & Marchal, L. Scanning electron microscopic study of Campylobacter pylori and associated gastroduodenal lesions [in French]. Gastroenterol. Clin. Biol. 13, 65B–70B (1989).
Amieva, M. R., Salama, N. R., Tompkins, L. S. & Falkow, S. Helicobacter pylori enter and survive within multivesicular vacuoles of epithelial cells. Cell. Microbiol. 4, 677–690 (2002).
Semino-Mora, C. et al. Intracellular and interstitial expression of Helicobacter pylori virulence genes in gastric precancerous intestinal metaplasia and adenocarcinoma. J. Infect. Dis. 187, 1165–1177 (2003).
Israel, D. A. et al. Helicobacter pylori genetic diversity within the gastric niche of a single human host. Proc. Natl Acad. Sci. USA 98, 14625–14630 (2001).
Philpott, D. J. et al. Reduced activation of inflammatory responses in host cells by mouse-adapted Helicobacter pylori isolates. Cell. Microbiol. 4, 285–296 (2002).
Azuma, T. et al. Association between diversity in the Src homology 2 domain-containing tyrosine phosphatase binding site of Helicobacter pylori CagA protein and gastric atrophy and cancer. J. Infect. Dis. 189, 820–827 (2004).
Aras, R. A. et al. Natural variation in populations of persistently colonizing bacteria affect human host cell phenotype. J. Infect. Dis. 188, 486–496 (2003).
Loughlin, M. F., Barnard, F. M., Jenkins, D., Sharples, G. J. & Jenks, P. J. Helicobacter pylori mutants defective in RuvC Holliday junction resolvase display reduced macrophage survival and spontaneous clearance from the murine gastric mucosa. Infect. Immun. 71, 2022–2031 (2003).
Wain, J. et al. Molecular typing of multiple-antibiotic-resistant Salmonella enterica serovar Typhi from Vietnam: application to acute and relapse cases of typhoid fever. J. Clin. Microbiol. 37, 2466–2472 (1999).
Levine, M. M., Black, R. E. & Lanata, C. Precise estimation of the numbers of chronic carriers of Salmonella typhi in Santiago, Chile, an endemic area. J. Infect. Dis. 146, 724–726 (1982).
Wain, J. et al. Quantitation of bacteria in bone marrow from patients with typhoid fever: relationship between counts and clinical features. J. Clin. Microbiol. 39, 1571–1576 (2001).
Sinnott, C. R. & Teall, A. J. Persistent gallbladder carriage of Salmonella typhi. Lancet 1 (8539), 976 (1987).
Monack, D. M., Bouley, D. M. & Falkow, S. Salmonella typhimurium persists within macrophages in the mesenteric lymph nodes of chronically infected Nramp1+/+ mice and can be reactivated by IFNγ neutralization. J. Exp. Med. 199, 231–241 (2004).
Vidal, S. M., Malo, D., Vogan, K., Skamene, E. & Gros, P. Natural resistance to infection with intracellular parasites: isolation of a candidate for Bcg. Cell 73, 469–485 (1993).
Merrell, D. S. & Camilli, A. Information overload: assigning genetic functionality in the age of genomics and large-scale screening. Trends Microbiol. 10, 571–574 (2002).
Koehler, J. E. in Persistent Bacterial Infections (eds Nataro, J. M. & Blaser, M. J.) 339–353 (ASM Press, Washington DC, 2000).
Koehler, J. E., Glaser, C. A. & Tappero, J. W. Rochalimaea henselae infection. A new zoonosis with the domestic cat as reservoir. J. Am. Med. Assoc. 271, 531–535 (1994).
Schulein, R. et al. Invasion and persistent intracellular colonization of erythrocytes. A unique parasitic strategy of the emerging pathogen Bartonella. J. Exp. Med. 193, 1077–1086 (2001).
Koehler, J. E., Quinn, F. D., Berger, T. G., LeBoit, P. E. & Tappero, J. W. Isolation of Rochalimaea species from cutaneous and osseous lesions of bacillary angiomatosis. N. Engl. J. Med. 327, 1625–1631 (1992).
Koesling, J., Aebischer, T., Falch, C., Schulein, R. & Dehio, C. Cutting edge: antibody-mediated cessation of hemotropic infection by the intraerythrocytic mouse pathogen Bartonella grahamii. J. Immunol. 167, 11–14 (2001).
Capo, C., Amirayan-Chevillard, N., Brouqui, P., Raoult, D. & Mege, J. L. Bartonella quintana bacteremia and overproduction of interleukin-10: model of bacterial persistence in homeless people. J. Infect. Dis. 187, 837–844 (2003).
Urban, B. C. et al. Plasmodium falciparum-infected erythrocytes modulate the maturation of dendritic cells. Nature 400, 73–77 (1999).
Schulein, R. & Dehio, C. The VirB/VirD4 type IV secretion system of Bartonella is essential for establishing intraerythrocytic infection. Mol. Microbiol. 46, 1053–1067 (2002).
Watarai, M., Makino, S. & Shirahata, T. An essential virulence protein of Brucella abortus, VirB4, requires an intact nucleoside-triphosphate-binding domain. Microbiology 148, 1439–1446 (2002).
Jenkins, M. K. et al. In vivo activation of antigen-specific CD4 T cells. Annu. Rev. Immunol. 19, 23–45 (2001).
Mascie-Taylor, C. G. & Karim, E. The burden of chronic disease. Science 302, 1921–1922 (2003).
Gubler, D. J. Resurgent vector-borne diseases as a global health problem. Emerg. Infect. Dis. 4, 442–450 (1998).
Hilbi, H. et al. Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. J. Biol. Chem. 273, 32895–32900 (1998).
Geng, Y. et al. Chlamydia pneumoniae inhibits apoptosis in human peripheral blood mononuclear cells through induction of IL-10. J. Immunol. 164, 5522–5529 (2000).
Danelishvili, L., McGarvey, J., Li, Y. J. & Bermudez, L. E. Mycobacterium tuberculosis infection causes different levels of apoptosis and necrosis in human macrophages and alveolar epithelial cells. Cell. Microbiol. 5, 649–660 (2003).
Braun, M. C., He, J., Wu, C. Y. & Kelsall, B. L. Cholera toxin suppresses interleukin (IL)-12 production and IL-12 receptor β1 and β2 chain expression. J. Exp. Med. 189, 541–552 (1999).
Vistica, B. P., McAllister, C. G., Sekura, R. D., Ihle, J. N. & Gery, I. Dual effects of pertussis toxin on lymphoid cells in culture. Cell. Immunol. 101, 232–241 (1986).
Mu, H. H. & Sewell, W. A. Enhancement of interleukin-4 production by pertussis toxin. Infect. Immun. 61, 2834–2840 (1993).
Thern, A., Stenberg, L., Dahlback, B. & Lindahl, G. Ig-binding surface proteins of Streptococcus pyogenes also bind human C4b-binding protein (C4BP), a regulatory component of the complement system. J. Immunol. 154, 375–386 (1995).
Grenier, D. Inactivation of human serum bactericidal activity by a trypsinlike protease isolated from Porphyromonas gingivalis. Infect. Immun. 60, 1854–1857 (1992).
Peterson, P. K., Verhoef, J., Sabath, L. D. & Quie, P. G. Effect of protein A on staphylococcal opsonization. Infect. Immun. 15, 760–764 (1977).
Bjorck, L. Protein L. A novel bacterial cell wall protein with affinity for Ig L chains. J. Immunol. 140, 1194–1197 (1988).
Stenger, S., Niazi, K. R. & Modlin, R. L. Down-regulation of CD1 on antigen-presenting cells by infection with Mycobacterium tuberculosis. J. Immunol. 161, 3582–3588 (1998).
Krall, R., Schmidt, G., Aktories, K. & Barbieri, J. T. Pseudomonas aeruginosa ExoT is a Rho GTPase-activating protein. Infect. Immun. 68, 6066–6068 (2000).
Barbieri, J. T. Pseudomonas aeruginosa exoenzyme S, a bifunctional type-III secreted cytotoxin. Int. J. Med. Microbiol. 290, 381–387 (2000).
Marra, A., Blander, S. J., Horwitz, M. A. & Shuman, H. A. Identification of a Legionella pneumophila locus required for intracellular multiplication in human macrophages. Proc. Natl Acad. Sci. USA 89, 9607–9611 (1992).
Berger, K. H. & Isberg, R. R. Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol. Microbiol. 7, 7–19 (1993).
Baca, O. G., Li, Y. P. & Kumar, H. Survival of the Q fever agent Coxiella burnetii in the phagolysosome. Trends Microbiol. 2, 476–480 (1994).
Gray-Owen, S. D., Dehio, C., Rudel, T., Naumann, M. & Meyer, T. F. in Principles of Bacterial Pathogenesis (ed. Groisman, E. A.) 559–618 (Academic Press, San Diego, 2001).
Acknowledgements
The authors thank J. Koehler, L. Thompson, A. Mueller and A. Camilli for comments on the manuscript. Research in the laboratories of D.S.M. and S.F. are supported by funds from the Damon Runyon Foundation and the National Institutes of Health.
Author information
Authors and Affiliations
Ethics declarations
Competing interests
The authors declare that they have no competing financial interests.
Rights and permissions
About this article
Cite this article
Merrell, D., Falkow, S. Frontal and stealth attack strategies in microbial pathogenesis. Nature 430, 250–256 (2004). https://doi.org/10.1038/nature02760
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature02760
This article is cited by
-
Effect of Helicobacter pylori and Helminth Coinfection on the Immune Response to Mycobacterium tuberculosis
Current Microbiology (2021)
-
IcmF and DotU are required for the virulence of Acidovorax oryzae strain RS-1
Archives of Microbiology (2018)
-
Characterization and differentiation of equine experimental local and early systemic inflammation by expression responses of inflammation-related genes in peripheral blood leukocytes
BMC Veterinary Research (2016)
-
Vasculitis, cerebral infarction and persistent Bartonella henselae infection in a child
Parasites & Vectors (2016)
-
Ecto-5′-Nucleotidase (CD73) Regulates Host Inflammatory Responses and Exacerbates Murine Salmonellosis
Scientific Reports (2014)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
