
Abstract
Many human cancers occur in renewing epithelial tissues, in which cellular lineages typically go through two distinct phases: early in life, cell populations expand exponentially to form the tissue, and for the remainder of life, the tissue is renewed by stem cells dividing to create an almost linear cellular history1. Here we use a simple mathematical model to show that mutations that arise during the exponential phase probably seed tissues with stem cells carrying mutations that may predispose to cancer. Susceptibility to late-life cancers, such as those of the skin and colon, may therefore be influenced by somatic mutations that occur during early development.
Similar content being viewed by others

Main
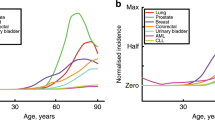
Mutations accumulate during exponential cellular growth according to the Luria–Delbrück distribution2. Let ue be the mutation probability per cell division during exponential growth, and N the number of stem cells produced during development to seed the tissue. Starting with one cell, the number of cell divisions required to produce N cells is N − 1, and the total number of mutation events during the cellular history is M = ue(N − 1) ≈ ueN. (The phases of cellular growth are shown in Fig. 1a.)

a, The phases of cellular growth in epithelial tissues. Cell populations increase exponentially during development, shown by a branching phase of division; at the end of development, stem cells differentiate in each tissue compartment (circles); stem cells renew each compartment by dividing to form a nearly linear cellular history (dashed lines) — each stem-cell division gives rise to one daughter stem cell that continues to renew the tissue and to one daughter transit cell that divides rapidly to produce a short-lived lineage that fills the tissue. b, The cumulative probability, p, for the number of stem cells mutated during development. By plotting log10[p/(1 − p)], the zero line gives the median of the distribution. N, number of stem cells produced during exponential growth. The number above each line is ue, the mutation probability per cell added to the population during exponential growth. For a single gene, ue is probably of the order of 10−7; thus, if there are 100 genes for which initial mutations can influence the progression to cancer, then ue is roughly of the order of 10−5. The Luria–Delbrück distribution plotted here was calculated using equation (8) of ref. 7.
Figure 1b shows the probability distribution for the number of stem cells that carry mutations. These are the mutations that arise during exponential growth, before further division of the stem lineages to renew the local tissue. The average frequency of stem cells with mutations is \(\overline{x}\) ≈ ueln(N) (ref. 2).
Although the frequency of stem cells that initially contain a mutation may be small, those mutations can contribute substantially to the total risk, RT, of cancer. Suppose that k rate-limiting mutations are needed for cancer to develop3,4, then the total risk is RT = N(1 − x)Rk + NxRk − 1, where x is the frequency of stem cells that start with one mutation, and Rk is the risk that a particular stem-cell lineage acquires k mutations during the phase of linear division and tissue renewal. The risk, Rk, is given roughly by the gamma distribution, which describes the probability of the occurrence of the kth event over a particular time interval. From the gamma distribution, Rk ≈ (usτ)k/k!, where us is the mutation rate per stem-cell division, and τ is the total number of stem-cell divisions.
The expected fractional increase in cancer risk arising from stem cells that begin with one mutation can be calculated as F = \(\overline{x}\)Rk − 1/Rk ≈ ueln(N)k/usτ. The next step is to assign approximate magnitudes to these quantities. We can take k ≈ 5 for the number of rate-limiting mutations required to cause epithelial cancer in humans3,4, ln(N) ≈ 20, and τ to range from 100–1,000. This gives the fractional increase in cancer risk, F, ranging from ue/10us to ue/us.
If mutations accumulate with the same probability per cell division during exponential growth and linear stem-cell division (ue = us), then the increase in risk from mutations arising in development ranges from 10–100%. If, as has been claimed5, mutation rates in stem cells are much lower than those during exponential growth, (us ≪ ue), then almost all cancer arises from predisposed stem-cell lineages that were mutated during development.
The risk of cancer from somatic mutations during development could be quantified by studying inbred rodents. The number of predisposing mutations per individual will vary according to the Luria–Delbrück distribution (Fig. 1b). Individuals with many predisposing mutations are likely to develop multiple independent tumours relatively early in life, whereas those with few predisposing mutations should develop few tumours relatively late in life. Controlled experiments have shown variability in cancer susceptibility among inbred rodents6, but the causes of this variability have not been determined.
Mutations during development seed a young individual with a small fraction of mutated stem cells. Those relatively few mutated lineages may therefore be responsible for a substantial proportion of the cancers that develop later in life.
References
Cairns, J. Nature 255, 197–200 (1975).
Zheng, Q. Math. Biosci. 162, 1–32 (1999).
Knudson, A. G. Proc. Natl Acad. Sci. USA 90, 10914–10921 (1993).
Hanahan, D. & Weinberg, R. A. Cell 100, 57–70 (2000).
Cairns, J. Proc. Natl Acad. Sci. USA 99, 10567–10570 (2002).
Prehn, R. T. Science 190, 1095–1096 (1975).
Zheng, Q. Math. Biosci. 176, 237–252 (2002).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Frank, S., Nowak, M. Developmental predisposition to cancer. Nature 422, 494 (2003). https://doi.org/10.1038/422494a
Issue Date:
DOI: https://doi.org/10.1038/422494a
This article is cited by
-
Chance, ignorance, and the paradoxes of cancer: Richard Peto on developing preventative strategies under uncertainty
European Journal of Epidemiology (2023)
-
Optimal solution of the fractional order breast cancer competition model
Scientific Reports (2021)
-
Hierarchical tissue organization as a general mechanism to limit the accumulation of somatic mutations
Nature Communications (2017)
-
Excess of mutational jackpot events in expanding populations revealed by spatial Luria–Delbrück experiments
Nature Communications (2016)
-
Risk factors and random chances
Nature (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
