
Abstract
The tripartite toxin secreted by Bacillus anthracis, the causative agent of anthrax, helps the bacterium evade the immune system and can kill the host during a systemic infection. Two components of the toxin enzymatically modify substrates within the cytosol of mammalian cells: oedema factor (OF) is an adenylate cyclase that impairs host defences through a variety of mechanisms including inhibiting phagocytosis1,2; lethal factor (LF) is a zinc-dependent protease that cleaves mitogen-activated protein kinase kinase and causes lysis of macrophages3,4,5. Protective antigen (PA), the third component, binds to a cellular receptor and mediates delivery of the enzymatic components to the cytosol. Here we describe the cloning of the human PA receptor using a genetic complementation approach. The receptor, termed ATR (anthrax toxin receptor), is a type I membrane protein with an extracellular von Willebrand factor A domain that binds directly to PA. In addition, a soluble version of this domain can protect cells from the action of the toxin.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others
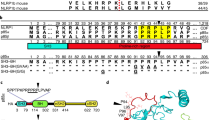

References
Leppla, S. H. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cAMP concentrations in eukaryotic cells. Proc. Natl Acad. Sci. USA 79, 3162–3166 (1982).
O'Brien, J. et al. Effects of anthrax toxin components on human neutrophils. Infect. Immun. 47, 306–310 (1985).
Duesbery, N. S. et al. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 280, 734–737 (1998).
Pellizzari, R. et al. Anthrax lethal factor cleaves MKK3 in macrophages and inhibits the LPS/IFNγ-induced release of NO and TNFα. FEBS Lett. 462, 199–204 (1999).
Friedlander, A. M. Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process. J. Biol. Chem. 261, 7123–7126 (1986).
Molloy, S. S. et al. Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen. J. Biol. Chem. 267, 16396–16402 (1992).
Milne, J. C. et al. Anthrax protective antigen forms oligomers during intoxication of mammalian cells. J. Biol. Chem. 269, 20607–20612 (1994).
Elliott, J. L., Mogridge, J. & Collier, R. J. A quantitative study of the interactions of Bacillus anthracis edema factor and lethal factor with activated protective antigen. Biochemistry 39, 6706–6713 (2000).
Gordon, V. M., Leppla, S. H. & Hewlitt, E. L. Inhibitors of receptor-mediated endocytosis block the entry of Bacillus anthracis adenylate cyclase toxin but not that of Bordetella pertussis adenylate cyclase toxin. Infect. Immun. 56, 1066–1069 (1988).
Blaustein, R. O., Koehler, T. M., Collier, R. J. & Finkelstein, A. Anthrax toxin: Channel-forming activity of protective antigen in planar phospholipid bilayers. Proc. Natl Acad. Sci. USA 86, 2209–2213 (1989).
Koehler, T. M. & Collier, R. J. Anthrax toxin protective antigen: low-pH-induced hydrophobicity and channel formation in liposomes. Mol. Microbiol. 5, 1501–1506 (1991).
Milne, J. C. & Collier, R. J. pH-dependent permeabilization of the plasma membrane of mammalian cells by anthrax protective antigen. Mol. Microbiol. 10, 647–653 (1993).
Escuyer, V. & Collier, R. J. Anthrax protective antigen interacts with a specific receptor on the surface of CHO-K1 cells. Infect. Immun. 59, 3381–3386 (1991).
Friedlander, A. M. et al. Characterization of macrophage sensitivity and resistance to anthrax lethal toxin. Infect. Immun. 61, 245–252 (1993).
Taft, S. A., Liber, H. L. & Skopek, T. R. Mutational spectrum of ICR-191 at the hprt locus in human lymphoblastoid cells. Environ. Mol. Mutagen. 23, 96–100 (1994).
Milne, J. C., Blanke, S. R., Hanna, P. C. & Collier, R. J. Protective antigen-binding domain of anthrax lethal factor mediates translocation of a heterologous protein fused to its amino- or carboxy-terminus. Mol. Microbiol. 15, 661–666 (1995).
Burns, J. C. et al. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc. Natl Acad. Sci. USA 90, 8033–8037 (1993).
Kitamura, T. et al. Efficient screening of retroviral cDNA expression libraries. Proc. Natl Acad. Sci. USA 92, 9146–9150 (1995).
Whitehead, I., Kirk, H. & Kay, R. Expression cloning of oncogenes by retroviral transfer of cDNA libraries. Mol. Cell. Biol. 15, 704–710 (1995).
St Croix, B. et al. Genes expressed in human tumor endothelium. Science 289, 1197–1202 (2000).
Dickeson, S. K. & Santaro, S. A. Ligand recognition by the I domain-containing integrins. Cell Mol. Life Sci. 54, 556–566 (1998).
Lee, J. O., Rieu, P., Arnaout, M. A. & Liddington, R. Crystal structure of the a domain from the subunit of integrin CR3 (CD11b/CD18). Cell 80, 631–638 (1995).
Molloy, S. S., Anderson, E. D., Jean, F. & Thomas, G. Bi-cycling the furin pathway: from TGN localization to pathogen activation and embryogenesis. Trends Cell Biol. 9, 28–35 (1999).
Beauregard, K. E., Wimer-Mackin, S., Collier, R. J. & Lencer, W. I. Anthrax toxin entry into polarized epithelial cells. Infect. Immun. 67, 3026–3030 (1999).
Petosa, C. et al. Crystal structure of the anthrax toxin protective antigen. Nature 385, 833–838 (1997).
Varughese, M., Teixeira, A. V., Liu, S. & Leppla, S. H. Identification of a receptor-binding region within domain 4 of the protective antigen component of anthrax toxin. Infect. Immun. 67, 1860–1865 (1999).
Ory, D. S., Neugeboren, B. A. & Mulligan, R. C. A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitus virus G pseudotypes. Proc. Natl Acad. Sci. USA 93, 11400–11406 (1996).
Snitkovsky, S. et al. A TVA-single-chain antibody fusion protein mediates specific targeting of a subgroup A avian leukosis virus vector to cells expressing a tumor-specific form of epidermal growth factor receptor. J. Virol. 74, 9540–9545 (2000).
Legge, G. B. et al. NMR solution structure of the inserted domain of human leukocyte function associated antigen-1. J. Mol. Biol. 295, 1251–1264 (2000).
Emsley, J., King, S. L., Bergelson, J. M. & Liddington, R. C. Crystal structure of the I domain from Integrin α2β1. J. Biol. Chem. 272, 28512–28517 (1997).
Mogridge, J., Mourez, M. & Collier, R. J. Involvement of domain 3 in oligomerization by the protective antigen moiety of anthrax toxin. J. Bacteriol. 183, 2111–2116 (2001).
Acknowledgements
We thank K. Schell and members of the flow cytometry facility at the University of Wisconsin Comprehensive Cancer Center for performing the cell sorting. This work was supported by a grant to J.A.T.Y. and R.J.C. from the National Institutes of Health. J.M. was supported in part by a Medical Research Council (Canada) postdoctoral fellowship.
Author information
Authors and Affiliations
Ethics declarations
Competing interests
R.J.C. holds financial interest in AVANT Immunotherapeutics and PharmAthene, Inc.
Rights and permissions
About this article
Cite this article
Bradley, K., Mogridge, J., Mourez, M. et al. Identification of the cellular receptor for anthrax toxin. Nature 414, 225–229 (2001). https://doi.org/10.1038/n35101999
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/n35101999
This article is cited by
-
Targeting the Inside of Cells with Biologicals: Toxin Routes in a Therapeutic Context
BioDrugs (2023)
-
Cancer cell survival depends on collagen uptake into tumor-associated stroma
Nature Communications (2022)
-
Capillary morphogenesis gene 2 (CMG2) mediates growth factor-induced angiogenesis by regulating endothelial cell chemotaxis
Angiogenesis (2022)
-
Anthrax toxins regulate pain signaling and can deliver molecular cargoes into ANTXR2+ DRG sensory neurons
Nature Neuroscience (2022)
-
Guide RNAs with embedded barcodes boost CRISPR-pooled screens
Genome Biology (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
