
Abstract
The draft human genome sequence is an important step in cataloguing the molecular hardware that supports the processes of life. Here I look at what we have learned from the draft sequence about our cytoskeletal and motility systems. Most cytoskeletal and motility proteins were discovered previously by biochemical isolation, traditional cloning methods or random sequences of complementary DNAs. The ongoing challenges of assembling and annotating genes for motor proteins with long, fragmented coding sequences emphasize the importance of expert knowledge of related proteins and confirmatory evidence from cDNA sequences.
Similar content being viewed by others

Main
Humans and other higher animals have three cytoskeletal systems: actin filaments, microtubules and intermediate filaments1. The myosin family of molecular motors pull on actin filaments or move cargo along them. Dynein and kinesin motors move microtubules or move cargo along microtubules.
How many genes?
Traditional biochemical and genetic methods have identified many protein components of these three systems: 6 mammalian actins and more than 70 families of actin-binding proteins; about 6 vertebrate α-tubulins and β-tubulins (forming the dimeric subunit of microtubules) and about a dozen families of microtubule-binding proteins; and about 31 human intermediate filament proteins and 5 families of associated proteins. Most families of proteins that bind one of the cytoskeletal polymers consist of several isoforms, resulting from multiple genes, alternative splicing or both. Divergent actins called actin-related proteins or Arps2 have special functions and contribute to the diversity of the system.
The draft genome sequence might have unveiled a cornucopia of new cytoskeletal genes. Instead, it appears to confirm that most of these genes were found by traditional methods. For example, the known actin family included six functional actin genes, more than twenty pseudogenes and six families of Arps encoded by nine genes (Table 1). The draft genome locates many of the pseudogenes and reveals at least fourteen new genes: seven highly divergent actin genes and seven new Arp genes. Work is required to establish whether these genes are functional. More genes may emerge, as two known actin genes are not recognized in the draft sequence.
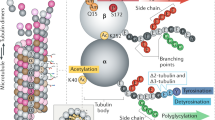
Genes for complex, multi-domain proteins such as the motor proteins myosin and kinesin are more challenging to assemble than genes for small, single-domain proteins such as profilin and actin (Fig. 1). However, researchers found virtually all of the genes for myosin and kinesin using traditional cloning methods and searches of cDNA databases. The current annotation of the draft human genome sequence includes most of those genes, but many are represented as fragments that have not yet been assembled into full-length coding sequences. The genes are huge, fragmented by introns, and include multiple non-homologous domains outside the core energy-transducing domains.

Variable N- and C-terminal domains are light blue; other colours represent core domains. Neither profilin nor actin has accessory domains. Myosin and kinesin genes have a range of variable domains at the N- and/or C-termini of the core ‘catalytic’ and ‘motor’ domains. Myosins have 1–7 IQ domains, which bind light chains or calmodulin.
Myosin and kinesin researchers found about 40 myosin genes3 and 40 kinesin genes (R. Freedman and K. Wood, personal communication). These were found initially by biochemical isolation, directed cloning and characterization. Searches of emerging cDNA and genomic databases completed the inventories of full-length genes. Myosin and kinesin each have their own defining catalytic domain, the major features of which remain in protein sequences even after many rounds of gene duplication and considerable sequence divergence. Sequence comparisons readily identify these catalytic domains, but finding the rest of these large genes is challenging. Most kinesins and myosins have a large carboxy-terminal tail consisting of many divergent domains (Fig. 1). Some have amino-terminal domains as well, and many introns fragment the genes. Nevertheless, an experienced worker with in-depth knowledge of the gene families can usually piece together complete genes by starting with a homologous catalytic domain and searching cDNA and genomic sequences in public databases for the flanking sequences.
Given the draft human sequence, annotators set out to predict coding sequences globally. The gene assembly procedures identified coding sequences for parts or all of many homologous catalytic domains of the motor proteins. A search of the contracted protein dataset of 17 July 2000 with the catalytic domain of a human cytoplasmic myosin returned about 80 coding sequences for myosins, but few correct, full-length coding sequences for myosin genes that had not already been defined by traditional methods. Few known myosin genes were missed, but owing to the fragmentary nature of the sequences on the current hit list, some hits are parts of the same gene. Thus the final number of myosin genes will be fewer than the current hits. The situation is similar for kinesins. Therefore, current estimates of the number of human genes for complex, multidomain proteins are likely to be inflated to some extent and must be considered to be provisional.
The WASp/Scar family represents the middle ground. These signal-transducing proteins regulate actin filament nucleation by the Arp2/3 complex4, and are less well characterized than myosins or kinesins. They are not large, but their multiple, incompletely conserved domains present challenges for gene assembly. All have WH2 domains (which bind an actin monomer), A domains (which bind Arp2/3) and proline-rich sequences (which bind profilin and SH3 domains). WASPs, but not Scars, also have WH1 domains (which bind WIP/verprolin) and GTPase-binding domains (which bind Cdc42). Traditional methods revealed human genes for WASp, N-WASP and three Scars (also called WAVE). The provisional human protein dataset includes several candidates for new WASp/Scar proteins, identified by sequence similarity to WH1, WH2/A or A domains (W.-L. Lee, personal communication). RNB6 appears to have a WH1 domain, but no WH2 or A domains. IGI_M1_ctg18730_17 and Q9Y6W5 have WH2 and A domains, but are gene fragments requiring further assembly, verification with cDNAs and testing for relevant activities.
These examples illustrate the importance of individual effort by biological specialists in completing the annotation of the human genome. Given inevitable sequencing errors and huge introns, accurate gene assembly from genomic sequences requires in-depth knowledge of related genes. Improving the completeness of collections of full-length cDNA sequences will be valuable in validating the annotation of genome sequences. The most constructive approach might be to develop guidelines and tools for annotating genes and gene families and then farm out most of the work to experts on each biological process, as suggested by Brinkman et al.5. This is one case where small science will yield a better product than the industrial approach required for sequencing.
Impact
Once completed, will the inventory of genes aid or distract from the search for general principles? For 50 years the model for understanding of biology has been to study a model system in depth and then to extrapolate its principles to related physiological systems. However, I know of no protein that has been understood in any depth without a decade or more of intense effort by laboratories devoted to determining its structure, interactions with partner molecules and roles in cellular physiology. Given finite resources, scientists cannot analyse every human gene product in such detail. I fear that the temptation to analyse full genome sequences will prove irresistible, perhaps even delaying the laboratory work required to complete the mechanistic analysis needed to understand physiology.
The complete genetic inventory will advance our understanding of disease. Although estimates vary, most human genes may contribute to disease. For example, genetic variants in each major contractile protein cause dysfunction of the human heart6. The same must be true in other organs. Thirty years ago the conditions caused by these mutations were called ‘idiopathic’ cardiomyopathies, and as recently as 1986 the idea that amino-acid substitutions in contractile proteins cause cardiomyopathies was still speculative.
The inventory will also reveal useful targets for development of new therapies. We now know, in principle, most of the potential drug targets that are available to treat human diseases. Of course, years of validation of these targets lies ahead, but the genome sequence provides limits. In the case of the cytoskeleton, this opportunity is untapped. Other than cancer drugs that bind microtubules (vinca alkaloids and taxol), no drugs in clinical use bind proteins of the cytoskeleton or molecular motors that operate our cardiovascular and musculo-skeletal systems.
The genetic inventory is essential to appreciate the complexity of the cytoskeleton, associated molecular motors and other systems. However, I doubt that the inventory of genes will provide much insight into molecular mechanisms, as even the simplest protein is multifaceted and has a complex mechanism of action. Genomics may help to identify networks of interacting proteins, but understanding how protein networks function requires details about the dynamics of the reaction pathways as well as the concentration and cellular localization of each component. Only continued work on atomic structures and the biophysics of molecular interactions and reactions, along with genetic or pharmacological tests for physiological functions, will reveal the mechanisms required to understand the essence of physiology and pathology.
References
Kreis, T. & Vale, R. (eds) Guidebook to the Cytoskeletal and Motor Proteins 2nd edn (Oxford Univ. Press, Oxford, New York, 1999).
Schroer, T. A. et al. Actin-related protein nomenclature and classification. J. Cell Biol. 127, 1777–1778 (1994).
Berg, J. S., Powell, B. C. & Cheney, R. E. A millennial census of the myosin superfamily. Mol. Biol. Cell (in the press).
Higgs, H. N. & Pollard, T. D. Regulation of actin polymerization by Arp2/3 complex and WASp/Scar proteins. J. Biol. Chem. 274, 32531–32534 (1999).
Brinkman, F. S. L., Hancock, R. E. W. & Stover, C. K. Sequencing solution: use volunteer annotators organized via Internet. Nature 406, 933 (2000).
Towbin, J. A. The role of cytoskeletal proteins in cardiomyopathies. Curr. Opin. Cell Biol. 10, 131–139 (1998).
Gunning, P. et al. Isolation and characterization of full length cDNA clones for human a-, β- and g-actin mRNAs: skeletal but not cytoplasmic actins have an amino-terminal cysteine that is subsequently removed. Mol. Cell. Biol. 3, 787–795 (1983).
Ng, S.-Y. et al. Evolution of the functional human β-actin gene and its multi-pseudogene family: conservation of noncoding regions and chromosomal dispersion of pseudogenes. Mol. Cell. Biol. 5, 2720–2732 (1985).
Schafer, D. A. & Schroer, T. A. Actin-related proteins. Annu. Rev. Cell. Dev. Biol. 15, 341–363 (1999).
Ueyama, H. . Inazawa, J., Nishino, H., Ohkubo, I. & Miwa, T. FISH localization of human cytoplasmic actin genes ACTB to 7p22 and ACTGT1 to 17q25 and characterization of related pseudogenes. Cytogenet. Cell Genet. 74, 221–224 (1996).
Acknowledgements
I thank E. Birney and A. Kasprzyk (European Bioinformatics Institute) for searching the human protein datasets for cytoskeletal proteins, and J. Berg and R. E. Cheney (Univ. North Carolina) and W.-L. Lee (Washington Univ.) for analysing these datasets.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pollard, T. Genomics, the cytoskeleton and motility. Nature 409, 842–843 (2001). https://doi.org/10.1038/35057029
Issue Date:
DOI: https://doi.org/10.1038/35057029
This article is cited by
-
Reciprocal regulation of cellular mechanics and metabolism
Nature Metabolism (2021)
-
Mining the draft human genome
Nature (2001)
-
Initial sequencing and analysis of the human genome
Nature (2001)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
