
Abstract
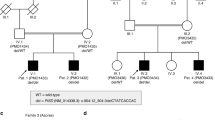
WAARDENGURG'S syndrome (WS) is an autosomal dominant combination of deafness and pigmentary disturbances, probably caused by defective function of the embryonic neural crest1,2. We have mapped one gene for WS to the distal part of chromosome 2 (ref. 3). On the basis of their homologous chromosomal location, their close linkage to an alkaline phosphatase gene, and their related phenotype, we suggested that WS and the mouse mutant Splotch4,5 might be homologous. Splotch is caused by mutation in the mouse Pax-3 gene6,7. This gene is one of a family of eight Pax genes known in mice which are involved in regulating embryonic development; each contains a highly conserved transcription control sequence, the paired box8. Here we show that some families with WS have mutations in the human homologue9 of Pax-3. Mutations in a related gene, Pax-6, which, like Pax-3, has both a paired box and a paired-type homeobox sequence, cause the Small-eye mutation in mice10 and aniridia inman11. Thus mutations in the Pax genes are important causes of human developmental defects.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others


References
Waardenburg, P. J. Am. J. hum. Genet. 3, 195–253 (1951).
Arias, S. Birth Defects 7, 87–101 (Williams & Wilkins, Baltimore, 1971).
Foy, C., Newton, V. E., Wellelley, D., Harris, R. & Read, A. P. Am. J. hum. Genet. 46, 1017–1023 (1990).
Auerbach, R. J. exp. Zool. 127, 305–329 (1954).
Epstein, D. J., Malo, D., Vekemans, M. & Gros, P. Genomics 10, 89–93 (1991).
Goulding, M. D., Chalepakis, G., Deutsch, U., Erselius, J. R. & Gruss, P. EMBO J . 10, 1135–1147 (1991).
Epstein, D. J., Vekemans, M. & Gros, P. Cell 67, 767–774 (1991).
Walther, C. et al. Genomics 11, 424–434 (1991).
Burri, M., Tromvoukis, Y., Bopp, D., Frigerio, G. & Noll, M. EMBO J. 8, 1183–1190 (1989).
Hill, R. E. et al. Nature 354, 522–525 (1991).
Ton, C. C. T. et al. Cell 67, 1059–1074 (1991).
Keen, J., Lester, D., Inglehearn, C., Curtis, A. & Bhattacharya, S. Trends Genet. 7, 5 (1991).
Steel, K. P. & Bock, G. R. Arch. Otolaryngol. 109, 22–29 (1983).
Asher, J. H. & Friedman, T. B. J. med. Genet. 27, 618–626 (1990).
Bopp, D., Burri, M., Baumgartner, S., Frigerio, G. & Noll, M. Cell 47, 1033–1040 (1986).
Deutsch, U., Dressler, G. R. & Gruss, P. Cell 53, 617–625 (1988).
Chalepakis, G. et al. Cell 66, 873–884 (1991).
Collier, S. et al. EMBO J. 8, 1393–1402 (1989).
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Tassabehji, M., Read, A., Newton, V. et al. Waardenburg's syndrome patients have mutations in the human homologue of the Pax-3 paired box gene. Nature 355, 635–636 (1992). https://doi.org/10.1038/355635a0
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/355635a0
This article is cited by
-
Waardenburg syndrome type 4 coexisting with open-angle glaucoma: a case report
Journal of Medical Case Reports (2022)
-
Genetic insights, disease mechanisms, and biological therapeutics for Waardenburg syndrome
Gene Therapy (2022)
-
Expression patterns of FSHD-causing DUX4 and myogenic transcription factors PAX3 and PAX7 are spatially distinct in differentiating human stem cell cultures
Skeletal Muscle (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
