
Abstract
Certain mutant Alzheimer’s amyloid-β (Aβ) peptides (that is, Dutch mutant APPE693Q) form complexes with gangliosides (GAβ). These mutant Aβ peptides may also undergo accelerated aggregation and accumulation upon exposure to GM2 and GM3. We hypothesized that increasing β-hexosaminidase (β-hex) activity would lead to a reduction in GM2 levels, which in turn, would cause a reduction in Aβ aggregation and accumulation. The small molecule OT1001 is a β-hex-targeted pharmacological chaperone with good bioavailability, blood–brain barrier penetration, high selectivity for β-hex and low cytotoxicity. Dutch APPE693Q transgenic mice accumulate oligomeric Aβ as they age, as well as Aβ oligomer-dose-dependent anxiety and impaired novel object recognition (NOR). Treatment of Dutch APPE693Q mice with OT1001 caused a dose-dependent increase in brain β-hex levels up to threefold over those observed at baseline. OT1001 treatment was associated with reduced anxiety, improved learning behavior in the NOR task and dramatically reduced GAβ accumulation in the subiculum and perirhinal cortex, both of which are brain regions required for normal NOR. Pharmacological chaperones that increase β-hex activity may be useful in reducing accumulation of certain mutant species of Aβ and in preventing the associated behavioral pathology.
Similar content being viewed by others

Introduction
Aberrant regulation of glycosphingolipids—specifically, gangliosides—has recently been associated with misfolding and aggregation of neurodegeneration-related proteins.1,2 Abnormal accumulation of gangliosides—which may act to seed these misfolding and aggregation events3,4—has been implicated as a key cellular factor in the pathogenesis of multiple diseases, including primary gangliosidoses (that is, Sandhoff, Tay–Sachs and Gaucher diseases). Ganglioside accumulation is also a feature of seemingly disparate diseases such as Niemann–Pick type C (NPC) disease and Alzheimer’s disease (AD). Beyond the accumulation of gangliosides, NPC and AD also share the development of similar structural neuropathology, including oligomerization and fibrilization of amyloid-β (Aβ) and tau.5 Conceivably, the gangliosidosis may be causative of the protein misfolding events shared between these seemingly very different neurodegenerative diseases.
The impact of neuronal glycosphingolipid accumulation as a driving force in neurodegenerative disease has also been highlighted by the discovery of a genetic association between Parkinson’s disease (PD) and Gaucher disease. Mutations in GBA1, encoding lysosomal glucocerebrosidase, are now recognized as an important genetic risk factor for PD.6 In support of this genetic link, it has been shown that decreased glucocerebrosidase activity is linked to α-synuclein accumulation in Gaucher and PD models. GBA knockdown in human iPS cells demonstrated a functional loss of glucocerebrosidase activity and accumulation of α-synuclein. In addition, wild-type glucocerebrosidase is inhibited by α-synuclein, thereby setting up a feed-forward loop that can drive progression of the neuropathology of PD.7,8
Similarly, growing evidence indicates that abnormalities in ganglioside metabolism may contribute to cerebral amyloidoses by accelerating the generation of neurotoxic forms of Aβ in the brain and/or cerebral vasculature. During aging and neurodegeneration, the physicochemical properties of membranes change, resulting in variable imbalances in the proportion of lipid subclasses in membranes, and these changes may contribute to the pathogenesis of AD.9, 10, 11, 12, 13, 14, 15 Consistent with this idea, elevated monosialoganglioside (GM1, GM2, GM3) levels have been reported in cerebral cortices from AD brains,16 where they appear to localize to membrane microdomains (detergent-resistant membranes).12,17 Ganglioside-bound Aβ (GAβ) peptide has been detected in brains that show only the earliest signs of AD pathology,18, 19, 20 suggesting that gangliosides may have some role(s) in initiating the pathogenesis of AD, such as the nucleation and/or seeding of Aβ oligomers and/or fibrils.21, 22, 23 Of note, some mutant forms of Aβ, especially those mutations that favor oligomerization over fibrillization (for example, Dutch APPE693Q), show a particular susceptibility to the pro-aggregation properties of GM2 and GM3.22,24
β-Hexosaminidase A (β-hex A) catabolizes GM2 ganglioside and its deficiency causes the autosomal recessive lysosomal storage disorders, such as Tay–Sachs disease and Sandhoff disease.25 We previously reported intraneuronal accumulation of Aβ-like immunoreactivity, α-synuclein-like immunoreactivity and phosphorylated tau-like immunoreactivity in the brains of HEXB KO mice.26 Biochemical and immunohistochemical analysis confirmed that the intraneuronal Aβ-like immunoreactivity represents APP‐CTFs (APP-C-terminal fragments) and/or Aβ but not full-length APP. In addition, we found increased levels of Aβ40 and Aβ42 peptides in the lipid-associated fraction relative to that recovered from wild-type brains. Accumulation of Aβ appeared to be associated with lysosomal-autophagic turnover of Aβ and fragments of APP that contain Aβ epitopes. Consistent with this finding, fibroblasts derived from Sandhoff and Tay–Sachs patients show significant accumulation of APP-CTFs, indicating that lysosomal dysfunction impairs APP-CTF clearance.27 Importantly, intraneuronal GAβ, a proposed prefibrillar or oligomeric aggregate found in AD, Aβ40 and Aβ42 immunoreactivity were observed in the brains of HEXB KO mice and throughout the frontal cortices of postmortem human GM1 gangliosidosis, Sandhoff disease and Tay–Sachs disease brains. Taken together, these results draw a link between the accumulation of gangliosides and the accumulation of Aβ.
GM2 and GM3 gangliosides have been shown to promote in vitro assembly of the Dutch-, Iowa- and Italian-type mutant Aβ peptides. All of these mutations are within the Aβ domain where they enhance oligomerization but largely preclude fibrillization.22,24 We previously reported the generation and characterization of Dutch APPE693Q transgenic mice that accumulate Aβ oligomers in the brain, with the highest levels observed within neurons in the subiculum, the visual cortex and the CA1 region of the hippocampus. These mice develop anxiety and learning behavior deficits, but, because of the Dutch mutation, the mice never develop amyloid deposits.28
Pharmacological chaperones (PCs) are small molecules that selectively bind and stabilize target wild-type proteins thereby facilitating proper folding, reducing premature degradation and increasing the efficiency of endoplasmic reticulum export.29,30 The small molecules are called ‘chaperones’ because they facilitate the movement of proteins from the compartment where they are synthesized (the endoplasmic reticulum) to that of their intended eventual residence (for example, the lysosome or the cell surface). Since these molecules are often based on inhibitors of their target protein, PCs must be reversible binders that facilitate proper trafficking and then, once the enzyme and PC arrive at their destination, the PC-enzyme complex dissociates so that the protein is free to carry out its proper function. This approach is broadly applicable to diseases where increasing the function or half-life of a specific protein is predicted to provide therapeutic benefit. PCs are in preclinical and clinical development for the treatment of numerous diseases including cystic fibrosis, retinitis pigmentosa, diabetes insipidus, PD and Fabry, Gaucher, Sandhoff and Tay–Sachs diseases.29,31, 32, 33 Amicus Therapeutics recently reported34 that, in a phase 3 clinical trial, a population of Fabry patients with disease due to particular amenable mutations demonstrated a statistically significant reduction in levels of kidney interstitial capillary GL-3 after 12 months of treatment with the PC migalastat HCl (that is, deoxygalactonojirimycin hydrochloride, DGJ HCl), an iminosugar PC that increases the activity of α-galactosidase A. Amicus Therapeutics has also assessed other iminosugar PCs including 1-deoxynojirimycin (1-DNJ) and isofagomine tartrate (IFG tartrate) for Pompe disease and Gaucher disease, respectively.29 In phase 1 and phase 2 studies, IFG tartrate significantly increased glucocerebrosidsase levels in peripheral blood mononuclear cells from healthy volunteers (phase 1) and type I Gaucher patients (phase 2); however, clinically meaningful end points were observed in only 1 of 18 Gaucher patients at the doses and dosing regimens assessed. Although these results were disappointing, the one patient who demonstrated a clinically meaningful improvement represents the first proof of concept in humans for the treatment of Gaucher disease by a PC therapy.
A PC targeting mutant forms of β-hex, a lysosomal enzyme that catabolizes GM2 gangliosides, has also been assessed in the clinic. Pyrimethamine, a non-carbohydrate PC for β-hex, was identified in an HTS screen of FDA-approved drugs.35 Pyrimethamine, like the iminosugar PC NAG-thiazoline,36 was shown to bind the active site of β-hex and rescue certain β-hex A mutants that cause Tay–Sachs disease or Sandhoff disease.37 In a phase 1/2 trial in Tay–Sachs and Sandhoff patients, pyrimethamine treatment increased β-hex A levels for all eight patients; however, severe side effects prevented further development.33 Carbohydrate mimetics including iminosugars like NAG-thiazoline are more potent and would likely demonstrate better tolerability. Three different iminosugars developed as PC therapies for Pompe disease, Gaucher disease and Fabry disease were shown to be well tolerated at all doses assessed in healthy volunteers (phase I studies).29 In addition, isofagomine and migalastat were shown to increase wild-type enzyme levels by two- to threefold for their respective targets, glucocerebrosidase and α-galactosidase A, in healthy volunteers. This surprising result highlights the fact that retention and premature degradation of incorrectly folded proteins is not restricted to mutant proteins. Schubert et al.38 have shown that large populations (up to 30%) of newly synthesized proteins are targeted for premature degradation by the proteasomes. PCs can increase cellular levels of many wild-type proteins by promoting protein folding, stability and/or endoplasmic reticulum export.30,39, 40, 41 Because several iminosugar PCs have been shown to rescue mutant forms of β-hex A and have been shown to be generally well tolerated in humans, we screened our own iminosugar library for compounds that interact and increase levels of wild-type β-hex (β-hex A/S/B) in cells and in the brains of C57Bl6 mice. Having identified a compound, OT1001, with good selectivity and in vivo pharmacokinetic properties, we hypothesized that our small molecule β-hex-targeted PC would increase brain β-hex A activity, thereby reducing GM2 levels, and, in turn, reducing Aβ oligomerization and behavioral pathology in Dutch APPE693Q transgenic mice.
In this study, we report that the β-hex-targeted PC OT1001 reduces GAβ pathology in Dutch APPE693Q transgenic mice and corrects their behavioral phenotype in a dose-dependent manner. We also show that OT1001 has good pharmaceutical qualities including oral bioavailability, brain penetration, tolerability, selectivity and low cytotoxicity. Thus, OT1001 itself is a viable drug candidate, and other strategies for increasing β-hex activity could be used for the treatment and management of Dutch CAA and other human cerebral Aβ amyloidoses.
Materials and methods
In vitro characterization of OT1001
Interaction of OT1001 with β-hex was demonstrated by inhibition of β-hex enzymatic activity. Wild-type fibroblast pellets (~2.5 × 106 cells per pellet) were prepared, frozen and then lysed with either pH 5 lysis buffer (82.4 mM sodium phosphate/58.8 mM citric acid, 0.25% sodium tauorocholate, 0.1% TX-100, pH 5) or pH 7 lysis buffer (100 mM sodium phosphate, 0.25% sodium tauorocholate, 0.1% TX-100, pH 7) and clarified by centrifugation. Fibroblast lysates were diluted 20:1 in pH 5 reaction buffer (82.4 mM sodium phosphate/58.8 mM citric acid, 0.25% sodium tauorocholate, pH 5) or pH 7 reaction buffer (100 mM sodium phosphate, 0.25% sodium tauorocholate, pH 7). In a 96-well plate, 10 μl of diluted fibroblast lysate was combined with 40 μl of 2.5 × OT1001 (final concentration 100 μM to 10 nM) and 50 μl of 4 mM of MUG (4-methylumbelliferyl N-acetyl-β-D-glucosaminide) substrate. Total β-hex activity was determined by incubation for 1 h at 37 °C and stopped with 100 μl of 0.5 M sodium carbonate. Released 4-methylumbelliferyl (4-MU) was measured using a Perkin Elmer Victor V fluorescence plate reader (Perkin Elmer, Waltham, MA, USA) using excitation of 355 nm and emission of 460 nm. Inhibition constants were calculated using the GraphPad Prism software package.
The ability of OT1001 to engage its intended target β-hex was assessed by treating human skin fibroblasts with increasing concentrations of OT1001 and measuring β-hex levels. Wild-type fibroblasts (CRL2076) were incubated with OT1001 (95–780 nM) for 5 days, followed by extensive washing with phosphate-buffered saline (5 × ), harvesting by trypsinization and centrifugation. Fibroblast pellets (~2.5–106 cells per pellet) were prepared, frozen and then lysed with pH 5 lysis buffer (82.4 mM sodium phosphate/58.8 mM citric acid, 0.25% sodium tauorocholate, 0.1% TX-100, pH 5) and clarified by centrifugation. Fibroblast lysates were diluted 20:1 in pH 5 reaction buffer (82.4 mM sodium phosphate/58.8 mM citric acid, 0.25% sodium tauorocholate, pH 5) for total β-hex assay. In a 96-well plate, 10 μl of diluted fibroblast lysate is combined with 90 μl of 2 mM final concentration of MUG substrate, incubated for 1 h at 37 °C and stopped with 100 μl of 0.5 M sodium carbonate. Released 4-MU was measured using a Perkin Elmer Victor V fluorescence plate reader using excitation of 355 nm and emission of 460 nm.
In vivo characterization of OT1001
OT1001 repeat dose/dose-response and PK studies
For single-dose PK studies, 5-week-old C57BL/J6 mice (n=5 mice per group, Taconic Farms, Germantown, NY, USA) received a single 100 mg kg−1 dose (oral gavage) of OT1001 and groups were euthanized at 0.5, 1, 2, 4, 8, 16, 24 and 48 h post dose for brain and plasma analyses of OT1001. For repeat dose PK studies, 5-week-old C57BL/J6 mice (n=5 mice per group, Taconic Farms, Germantown, NY, USA) were administered vehicle (water) or OT1001 (30, 100 or 300 mg kg−1) via oral gavage every day for 14 days. On day 15, mice were euthanized and brains were collected for analysis of β-hex levels. Briefly, whole brains were collected, flash frozen in liquid nitrogen and stored a −80 °C until analysis. Brain hemispheres were finely chopped with a razor blade and separated into 10–15 mg aliquots. An aliquot of whole brain tissue was homogenized in pH 5 lysis buffer (82.4 mM sodium phosphate/58.8 mM citric acid, 0.25% sodium tauorocholate, 0.1% TX-100, pH 5) and clarified by centrifugation (10 min, 4 °C at 20 000 g). Whole brain lysates were diluted 20:1 in pH 5 reaction buffer (82.4 mM sodium phosphate/58.8 mM citric acid, 0.25% sodium tauorocholate, pH 5) for all β-hex assays. In a 96-well format, 10 μl of diluted brain lysate was combined with 90 μl of a 4-MU substrate, incubated for 1 h at 37 °C, and the reaction was stopped with 100 μl of 0.5 M sodium carbonate. Released 4-MU was measured using a Perkin Elmer Victor V fluorescence plate reader using excitation of 355 nm and emission of 460 nm. Measurement of total β-hex activity and inhibition of total β-hex activity by OT1001 was carried out using 2 mM final concentration MUG; for β-hex A/S activity and inhibition, 2 mM of 4-methylumelliferyl N-acetyl-β-D-glucosamine-6-sulfate (MUGS); β-hex B activity was calculated as the difference between total β-hex and β-hex A/S activity. OT1001 levels were measured in plasma and brain samples using liquid chromatography-tandem mass spectrometry.
OT1001 study in Dutch APPE693Q transgenic mice
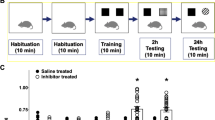
Animals and study design: We previously reported the generation and characterization of Dutch APPE693Q mice transgenic mice.28 Three-month-old male Dutch APPE693Q mice transgenic mice were subjected to oral gavage 5 days per week with either vehicle (water) (n=15) or 3, 10, 30 or 100 mg kg−1 OT1001 for 3 months (n=13). All animal studies were conducted in accordance with National Institute of Health Guidelines for the Care and Use of Experimental Animals and approved by the Institutional Animal Care and Use Committee at the Icahn School of Medicine at Mount Sinai. Mice were kept in a pathogen-free environment on a 12-h light/dark cycle, and given ad libitum access to food and water. At 6 months of age, all treatment groups, as well as a 3-month-old untreated baseline group (n=15), were cognitively assessed in the novel object recognition (NOR), elevated plus maze and fear conditioning (for fear conditioning see Supplementary Methods) tests. At 48 h following the final dose, mice were perfused with ice-cold 1 × phosphate-buffered saline. The brain was removed and dissected into two hemispheres. One half was snap-frozen on dry ice and stored at −80 °C for biochemistry (see Supplementary Methods for Aβ assay); the other half was post-fixed in 4% paraformaldehyde, then vibratome-sectioned in the sagittal plane into 30 μm sections and stored at 4 °C in storage buffer until histological analyses.
Behavioral testing: Mice were placed in the testing room 1 h before testing to acclimatize to the room. All testings were completed between 0800 and 1500 hours Tuesday through Friday, 2 weeks before final dosing of OT1001. All equipments were cleaned between animals.
Novel object recognition: On day 1, the mouse was habituated in the NOR arena (20-cm diameter) for 10 min. On day 2, the mouse underwent the testing phase, composed of two stages. During testing phase 1, the mouse was placed in the arena and allowed to explore two identical unfamiliar objects for 10 min. The mouse was then returned to its home cage for an interval of 1 h. During this time, one of the two objects that the mouse was previously allowed to explore was removed and replaced with a novel object. During testing phase 2, the mouse was placed back into the arena and allowed to explore the familiar object and the novel object for 4 min. Trials were videotaped using an overhead camera. The duration spent exploring the objects was then measured using ANY-maze (Stoelting, Wood Dale, IL, USA). Exploration was defined as the amount of time the mouse spent pointing its nose within 2 cm of the object.
Elevated plus maze: The maze was constructed in the configuration of a ‘plus’ sign, comprising two open arms (35 × 5 × 0 cm) across from each other and perpendicular to two closed arms (35 × 5 × 16 cm) with a central neutral zone (Stoelting). Each mouse, in turn, was placed in the neutral zone and allowed to explore for 5 min. Each trial was videotaped with an overhead camera and scored for the amount of time spent in the open and closed arms using ANY-maze (Stoelting). Entry into an arm was defined as having head and two paws into arms.
Histology: Aβ was assessed via free-floating immunohistochemistry using mAb 6E10 (1:1000, Covance, Princeton, NJ, USA) as previously described.28 Briefly, sections were incubated in 0.6% H2O2 in phosphate buffer for 30 min, then formic acid for 15 min, permeabilized with Triton X-100 in tris-buffered saline for 15 min, blocked with 3% goat serum in tris-buffered saline for 30 min, then incubated with mAb 6E10 (1:1000, Covance) overnight at 4 °C. The following day, sections were incubated with anti-mouse secondary antibody (1:200, Vector Laboratories, Burlingame, CA, USA) for 1 h. An ABC peroxidase kit and 3,3'-diaminobenzidine substrate kit (Vector Laboratories) were used to detect the biotinylated secondary antibody. Sections were then mounted and allowed to air dry before undergoing sequential dehydration with 10 min incubations in 50, 70 and 95% ethanol, followed by two 20-min incubations in 100% ethanol, 15-min incubation in 50% ethanol/50% xylene, a quick dip in 100% xylene and a final 15-min incubation in xylene, then cover slipped with DePeX (EMS, Hatfield, PA, USA).
GAβ was assessed via free-floating fluorescence immunohistochemistry using mouse anti-GAβ, clone 4396C (1:20), as previously described.26 Briefly, sections were permeabilized with Triton X-100 in tris-buffered saline for 30 min, blocked with 3% goat serum in tris-buffered saline for 30 min, then incubated with mouse anti-GAβ, clone 4396C (1:20) overnight at 4 °C. The following day sections were incubated with red fluorescent Alexa Fluor 594 (Invitrogen; 1:500) at room temperature for 1 h and mounted with Vectashield (Vector Laboratories). Images were captured on an Olympus BX61 upright microscope with an attached Olympus DP71 camera. Integrated density of GAβ staining was measured using Image J (National Institute of Health, Bethesda, MD, USA).
Biochemistry: Hexosaminidase activity—Brain hemispheres were finely chopped with a razor blade and separated into 10–15 mg aliquots. An aliquot of whole brain tissue was homogenized in pH 5 lysis buffer (82.4 mM sodium phosphate/58.8 mM citric acid, 0.25% sodium tauorocholate, 0.1% Triton X-100, pH 5) and clarified by centrifugation (10 min, 4 °C at 15 000 r.p.m.). Whole brain lysates were diluted 20 : 1 in pH 5 reaction buffer (82.4 mM sodium phosphate/58.8 mM citric acid, 0.25% sodium tauorocholate, pH 5). In a 96-well plate, 10 μl of diluted brain lysate is combined with 90 μl of a 4-MU substrate, incubated for 1 h at 37 °C and the reaction was stopped with 100 μl of 0.5 M sodium carbonate. Released 4-MU was measured using a Perkin Elmer Victor V fluorescence plate reader using excitation of 355 nm and emission of 460 nm. Measurement of β-hex A/S activity was carried out using 2 mM of MUGS; β-hex B activity was calculated as the difference between total β-hex and β-hex A/S activity.
Statistical analyses of behavioral, biochemical and histological outcomes—All data are presented as the mean±s.e.m. Statistical significance (P<0.05) was determined using Student’s t-tests or one-way analysis of variance with Bonferroni post hoc analyses (GraphPad Prism, San Diego, CA, USA).
Results
OT1001 is an iminosugar PC for β-hex
OT1001 is a small molecule (MW<500) PC that was identified by screening a library of ~500 iminosugars for inhibitors of β-hex and other lysosomal enzymes at pH 7. OT1001 is highly soluble in water (approximately 100 mg ml−1), has a predicted pKa of 7.85 and meets Lipinski’s rule of 5. OT1001 has been synthesized on a multi-gram scale, and it is anticipated that OT1001 could be synthesized in kilogram scale batches.
In vitro, OT1001 was a potent and specific inhibitor of β-hex (Ki=253.4 nM at pH 5; Ki=342.3 nM at pH 7; Figure 1a) and did not inhibit O-GlcNAcase (Supplementary Figures 1a and b) at concentrations up to 100 μM, or the lysosomal enzymes β-glucocerebrosidase (GCase), α-galactosidase (α-Gal) and β-galactosidase (β-Gal) at 100 μM, the highest concentration tested (Supplementary Figure 1c). In addition, OT1001 had little or no effect on cell viability for SH-SY5Y and human skin fibroblasts treated with concentrations as high as 1 mM OT1001 for up to 120 h (Supplementary Figures 1d and e). Treatment of healthy human skin fibroblasts for 5 days with 95, 390 or 780 nM OT1001 led to an increase in β-hex levels in a dose-dependent manner up to a maximum of threefold over baseline (Figure 1b), demonstrating the ability of OT1001 to engage its intended target, β-hex.

Characterization of β-hex-targeted pharmacological chaperone OT1001. OT1001 is a potent and specific inhibitor of β-hex demonstrating target engagement at sub-micromolar concentrations (a). OT1001 treatment of healthy human derived fibroblasts with OT1001 (95–780 nM) for 5 days resulted in a dose-dependent increase in wild-type total β-hex levels up to threefold (b). Five-week-old male C57BL/J6 mice were given a single 100 mg kg−1 dose of the β-hex-targeted pharmacological chaperone OT1001; plasma (c) and brain (d) OT1001 profiles. A 100 mg kg−1 dose of OT1001 crossed the blood–brain barrier and reached levels in the brain that are predicted to be sufficient to bind and increase β-hex levels (>342 nM) and dropped below Ki (253 nM at pH 5) levels for β-hex within 16 h (n=5 mice per time point). Five-week-old male C57BL/J6 mice were orally gavaged either with vehicle, 30, 100 or 300 mg kg−1 of the β-hex-targeted pharmacological chaperone OT1001 daily up to 14 days. Whole brain β-hex A&S (e) and β-hex B (f) activity (n=5 mice per time point). Data expressed as mean±s.e.m. β-hex, β-hexosaminidase.
OT1001 crosses the BBB and increases β-hex without obvious toxicity
To determine the appropriate doses and dosing regimens in the initial proof-of-concept study, we completed pharmacokinetic and dose-response/time course studies for OT1001 using C57BL/J6 mice (Figures 1c–f). The objectives of these studies were to confirm that OT1001 crosses the blood–brain barrier (BBB) and to determine the dose and minimum duration of daily dosing that provided the largest increase in brain levels of endogenous wild-type β-hex in C57BL/J6 mice, while simultaneously allowing OT1001 to intermittently drop below inhibitory concentrations (Ki at pH 5~253 nM) to enable disassociation from β-hex and to permit the turnover of substrate. In oral gavage, single-dose pharmacokinetic studies, we found that OT1001 (100 mg kg−1) crossed the BBB and reached levels in the brain that are predicted to be sufficient for binding to β-hex in the neutral pH environment of the endoplasmic reticulum (342 nM, OT1001’s Ki for β-hex inhibition at pH 7). The maximum concentration of OT1001 measured in the brain (Cmax,brain) was 1 μM, and the maximum plasma concentration (Cmax,plasma) was 100 μM, thereby yielding a brain-to-blood exposure (area under the curve ratio) of 4%. The plasma half-life of OT1001 was 1.3 h and cleared the brain at a slower rate with a brain half-life of 5 h. A brain half-life of 5 h allows OT1001 to clear for ~5 half-lives over 24 h; therefore, accumulation of drug in the brain should be minimal over the course of a 5-day dosing period, followed by a 72-h washout. The level of OT1001 dropped below that predicted to inhibit lysosomal β-hex (that is, 253 nM, OT1001’s Ki for β-hex at pH 5) within 16 h (Figures 1c and d). In addition, daily dosing of OT1001 for 14 days at 30, 100 or 300 mg kg−1 via gavage demonstrated a dose-dependent increase in β-hex level (Figures 1e and f), without increasing GM2 levels (not shown). At these doses, the maximum increase in β-hex level occurred at ~5 days of treatment (Figures 1e and f). As the half-life of elevated β-hex levels in the brain was ~2 days (data not shown), we chose the strategy of treating mice once daily for 5 days to maximize β-hex enhancement followed by 2 days without drug to maximize substrate turnover in the absence of OT1001. All doses were well tolerated by the mice.
OT1001 increases β-hex levels in Dutch APPE693Q transgenic mice
We next performed a proof-of-concept study to evaluate whether OT1001 reduced GAβ pathology in Dutch APPE693Q transgenic mice. First, we sought to determine whether OT1001 engaged its target β-hex in Dutch APPE693Q transgenic mice. We observed that a 3-month course of OT1001 increased brain β-hex (both B and A&S) in a dose-dependent manner up to threefold (Figures 2a and b). Thin layer chromatographic analysis of whole brain extracts did not reveal any changes in ganglioside levels, indicating that β-hex was not generally inhibited throughout the brain. However, regional changes could be masked as only whole total brain homogenates were assayed. The most significant reductions in GAβ were observed in the subiculum and perirhinal cortex regions, which would account for a very small percentage of the total brain volume used to generate the homogenates.

Dose-dependent increase in brain β-hex levels following treatment of β-hex-targeted pharmacological chaperone OT1001 in Dutch APPE693Q transgenic mice. Three-month-old male Dutch APPE693Q transgenic mice were either untreated, or orally dosed with vehicle or OT1001 for 3 months. Treatment of OT1001 increased β-hex B (a) and A&S (b) levels in a dose-dependent manner (untreated; n=3, vehicle; n=6, 3 and 10 mg kg−1 OT1001; n=7; for 30 and 100 mg kg−1 OT1001, n=6). Data are expressed as mean±s.e.m. One-way analysis of variance with Bonferroni post hoc analyses, *P<0.05, ***P<0.001. β-hex, β-hexosaminidase.
OT1001 prevents the appearance of the behavioral phenotype of Dutch APPE693Q transgenic mice
In the elevated plus maze test, we observed that 6-month-old vehicle-treated Dutch APPE693Q transgenic mice displayed more anxiety-like behavior than 3-month-old untreated mice (Figure 3a) as assessed by increased time spent in closed arms. We also assessed memory by employing the NOR test. Three-month-old untreated Dutch APPE693Q transgenic mice displayed intact NOR as evidenced by their spending significantly more time exploring the novel object over the familiar object, whereas 6-month-old vehicle-treated mice exhibited impaired NOR (Figure 3b).

Reduced anxiety behavior and prevention of onset of learning behavior deficits following treatment of the β-hex-targeted pharmacological chaperone OT1001 in Dutch APPE693Q transgenic mice. Three-month-old male Dutch APPE693Q transgenic mice were either untreated (n=15), or orally dosed with vehicle (n=15) or OT1001 (3, 10, 30 or 100 mg kg−1 OT1001; n=13 per treatment group) for 3 months. Six-month-old vehicle-treated Dutch APPE693Q transgenic mice show increased anxiety relative to 3-month-old untreated mice in the elevated plus maze, which is reversed following treatment with OT1001 at all doses tested (a). Three-month-old untreated mice display intact memory, exploring the novel (new) object more than the familiar (old) object, whereas 6-month-old vehicle-treated Dutch transgenic mice show a learning behavior deficit in novel object recognition (NOR); OT1001 prevents onset of learning deficit in the NOR test in a dose-dependent manner (b). Data expressed as mean±s.e.m. *P<0.05, ***P<0.001. β-hex, β-hexosaminidase.
A 3-month course of OT1001 was associated with reduced anxiety-like behavior in the elevated plus maze at all doses tested (Figure 3a) and protection from the typical aging-related memory deficits in the NOR test, at the higher doses (Figure 3b). Six-month-old Dutch APPE693Q transgenic mice treated with OT1001 at doses of 30 or 100 mg kg−1 displayed NOR of mice half their age. In contrast, OT1001 had no effect on contextual or cued fear conditioning (Supplementary Figure 2).
OT1001 reduces Aβ and GAβ pathology in Dutch APPE693Q transgenic mice
Next, we measured Aβ and GAβ in Dutch APPE693Q transgenic mice following a 3-month course of OT1001. In whole hemisphere homogenates, we found no significant (P>0.05, one-way analysis of variance with Bonferroni post hoc analyses) change in Aβ40 (Supplementary Figures 3a–d), Aβ42 (Supplementary Figures 3e–h), Aβ42/40 ratio (Supplementary Figures 3i–l), prefibrillar Aβ (Supplementary Figure 3m) or Aβ oligomer levels (Supplementary Figures 3n and o) at any dose of OT1001 tested. We then went on to look for regional changes in Aβ and/or GAβ. Immunohistochemical analysis revealed a qualitative region-specific reduction of Aβ accumulation, especially within the subiculum (Figures 4a and b). No differences were observed in the visual cortex (Figures 4c and d) nor in the CA1 region of the hippocampus (Figures 4e and f) of Dutch APPE693Q transgenic mice dosed with 100 mg kg−1 OT1001.

Aβ-like immunoreactivity was reduced following treatment of the β-hex-targeted pharmacological chaperone OT1001 in Dutch APPE693Q transgenic mice. Sagittal brains sections from male Dutch APPE693Q transgenic mice orally dosed either vehicle (a, c, e) or 100 mg kg−1 OT1001 (b, d, f) for 3 months were stained with the antibody 6E10 (vehicle; n=4, 100 mg kg−1 OT1001; n=4). Aβ immunoreactivity was less intense in the subiculum (a, b) but not visual cortex (c, d) nor in CA1 region of the hippocampus (e, f) of OT1001 treated Dutch APPE693Q transgenic mice. Scale bar, (a–f) 200 μM. β-hex, β-hexosaminidase.
GAβ was assessed via free-floating immunohistochemistry using mouse anti-GAβ, clone 4396C. Region-specific reduction in Aβ was accompanied by a striking quantitative reduction in GAβ within the subiculum (Figures 5a, b and k) and perirhinal cortex (Figures 5c, d and l), two areas in the brain implicated in the successful performance of the NOR test. The trend toward reduction in GAβ was observed in the lateral entorhinal cortex (Figures 5e, f and m), another brain region involved in NOR. A reduction in GAβ was observed but did not reach significance in the visual cortex (Figures 5g, h and n) or in the CA1 region of the hippocampus (Figures 5i, j and o). These results indicate that OT1001 treatment can have a regional effect on reducing GAβ levels without disrupting or altering normal Aβ processing throughout the brain.

GAβ immunoreactivity was reduced following treatment of the β-hex-targeted pharmacological chaperone OT1001 in Dutch APPE693Q transgenic mice. Sagittal brains sections from male Dutch APPE693Q transgenic mice dosed either vehicle (a, c, e, g, i) or OT1001 (b, d, f, h, j) for 3 months were stained with anti-GAβ (4396C) (vehicle; n=4, 100 mg kg−1 OT1001; n=4). GAβ-like immunoreactivity as detected by 4396C antibody was reduced in the subiculum and perirhinal cortex (k, l). GAβ was unchanged in the visual cortex, lateral entorhinal cortex and CA1 region of the hippocampus (m, n, o). Scale bar, (a–j) 200 μM. GAβ staining in vessels was detectable in the cortical regions (p). Data expressed as mean±s.e.m. Student’s t-tests, *P<0.05. β-hex, β-hexosaminidase; GAβ, ganglioside-bound Aβ.
Discussion
In this study, we demonstrated that the β-hex-targeted small molecule PC OT1001 has good bioavailability, good BBB penetration, high selectivity for β-hex and low cytotoxicity. We assessed the effect of OT1001 on Aβ and GAβ deposition and on anxiety-like behavior and NOR memory deficits in Dutch APPE693Q transgenic mice. We confirmed our previous findings that Dutch APPE693Q transgenic mice show aging-dependent deficits in anxiety-like behavior and NOR memory.28 Dutch APPE693Q transgenic mice had intact memory in the NOR test at 3 months of age, whereas 6-month-old vehicle-treated mice showed impaired NOR. The functional impairments observed in Dutch APPE693Q transgenic mice at 6 months of age were associated with regions most involved by Aβ accumulation: that is, the subiculum, the visual cortex and CA1 region of the hippocampus. As with our other studies, the strongest Aβ-like immunoreactivity was observed in the subiculum.28 Recent studies have implicated the subiculum in memory, specifically in the successful performance of the NOR test.42 We went on to observe that a β-hex-targeted PC corrects two features of the behavioral phenotype of the Dutch APPE693Q transgenic mouse in association with an apparent reduction of Aβ and GAβ accumulation. A 3-month course of OT1001 was associated with a threefold increase in β-hex levels in the brains of Dutch APPE693 transgenic mice, demonstrating that OT1001 was engaging its target. A striking quantitative reduction in GAβ was observed within the subiculum and perirhinal cortex, areas in the brain involved in NOR. A decrease in GAβ was also observed in the lateral entorhinal cortex, visual cortex and CA1 region of the hippocampus, but these changes did not reach statistical significance. As OT1001 had no significant effect on levels of Aβ40 and Aβ42, or Aβ40/42 ratios in whole brain extracts, OT1001 appears to lower GAβ and correct behavior deficits in the Dutch APP transgenic mice without causing a major general disruption of normal Aβ processing.
GAβ peptide has been detected in brains that show only the earliest signs of AD pathology,18, 19, 20 suggesting that gangliosides may have some role(s) early in the pathogenesis of AD, such as the nucleation or seeding of Aβ fibrils.21, 22, 23 Although the exact mechanism via which gangliosides ‘seed’ Aβ oligomerization to form fibrils remains unknown, several studies have provided interesting clues. Utsumi et al.43 showed that upon binding to gangliosidic micelles, Aβ40 exhibited an ‘up-and-down’ topology, where the C-terminal dipeptide segment and two α-helices were in contact with the hydrophobic interior, whereas the remaining regions were exposed to the aqueous environment.44 This interaction can induce Aβ oligomerization and fibril formation by promoting a change in Aβ conformation from α-helix to β-sheet.45,46 Interestingly, cholesterol has been shown to enhance binding of Aβ to GM1 ganglioside clusters in lipid rafts providing a potential mechanism for endosomal Aβ accumulation in Nieman Pick type C, where genetic mutations disrupt the function of the lysosomal transporter protein NPC1 leading to the accumulation of cholesterol and glycolipids.45,47 In addition to promoting binding and oligomerization, GM1 gangliosides have been shown to promote the sequestration of Aβ dimers onto membranes.48 Studies with GM2/GD2 synthase KO mice showed that the amount of dimers recovered from membrane fractions generally increased with increasing amounts of GM1 in the brain. In addition, GM1-Aβ was identified in the CSF of healthy individuals and correlated with Aβ42 levels, suggesting its potential use as a biomarker. GM1 gangliosides have received the most attention; however, Aβ has been shown to bind to a broad range of gangliosides including all major gangliosides and GM2.49 Therefore, it is not surprising that GAβ was found to accumulate in brains from patients with GM1 and GM2 gangliosidoses. These findings suggest that ganglioside clusters may serve as a unique platform for binding of Aβ and the promotion of and transitioning the specific intermolecular interactions.43,44 Therefore, increasing the activity of enzymes involved in ganglioside catabolism provide a novel approach to treating cerebral amyloidosis and other proteinopathies.
The notion of ganglioside-promoted Aβ-assembly is further supported by the observation that an antibody raised against ganglioside GM1-bound Aβ is sufficient to suppress Aβ deposition in a mouse AD model (Tg2576).50 The antibody also recognizes other gangliosides bound to variant-type Aβs, including GM1-, GM3- and GD3. GM2–Aβ complexes have not been assessed. Gangliosides might modulate regional Aβ deposition as they are expressed in a region-specific manner in the brain. Yamamoto et al.22,24 have shown that assembly of hereditary variant Dutch- and Iowa-type Aβ was accelerated by GM2 or GM3 gangliosides. Notably, Dutch- and Iowa- type Aβ are deposited in the cerebral vessel wall and cerebrovascular smooth muscle cells, both of which exclusively express GM2 and GM3.22 Thus, the assembly of hereditary Aβ variants may be accelerated by local environmental factors, including the distribution of particular gangliosides.22
In conclusion, our studies provide the first evidence that a β-hex-targeted PC increases wild-type β-hex levels while decreasing GAβ pathology in regions that demonstrate the highest amount of Aβ immunoreactivity. These molecular and histological changes occur in association with correction of two aspects of the behavioral phenotype in Dutch APPE693Q transgenic mice. These data could have implications for management of the human cerebral amyloidoses, particularly those associated with certain APP/Aβ mutations, such as Dutch CAA. OT1001 has good bioavailability, good BBB penetration, high selectivity for β-hex and low cytotoxicity making OT1001 a viable drug candidate. OT1001 can be readily advanced into clinical trials in patient populations with the Dutch APPE693Q mutation. The next steps for the development of OT1001 for this use will focus on completing IND-enabling studies and additional animal models that may identify other patient populations that might benefit from treatment with OT1001. Assessment of in vivo target engagement in human brain will also be a key next step.
References
Alcalay RN, Caccappolo E, Mejia-Santana H, Tang M, Rosado L, Orbe Reilly M et al. Cognitive performance of GBA mutation carriers with early-onset PD: the CORE-PD study. Neurology 2012; 78: 1434–1440.
Daniele A, Albanese A . Early visual memory deficits: a neuropsychological marker of GBA mutations in PD? Neurology 2012; 78: 1372–1373.
Lee H, Lee JK, Bae YC, Yang SH, Okino N, Schuchman EH et al. Inhibition of GM3 synthase attenuates neuropathology of niemann-pick disease type C by affecting sphingolipid metabolism. Mol Cells 2014; 37: 161–171.
Zervas M, Somers KL, Thrall MA, Walkley SU . Critical role for glycosphingolipids in Niemann-Pick disease type C. Curr Biol 2001; 11: 1283–1287.
Malnar M, Hecimovic S, Mattsson N, Zetterberg H . Bidirectional links between Alzheimer's disease and Niemann-Pick type C disease. Neurobiol Dis advance online publication, 4 June 2014; doi:10.1016/j.nbd.2014.05.033(e-pub ahead of print).
Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 2009; 361: 1651–1661.
Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011; 146: 37–52.
Yap TL, Velayati A, Sidransky E, Lee JC . Membrane-bound alpha-synuclein interacts with glucocerebrosidase and inhibits enzyme activity. Mol Genet Metab 2013; 108: 56–64.
Yanagisawa K . Role of gangliosides in Alzheimer's disease. Biochim Biophys Acta 2007; 1768: 1943–1951.
Mutoh T, Hirabayashi Y, Mihara T, Ueda M, Koga H, Ueda A et al. Role of glycosphingolipids and therapeutic perspectives on Alzheimer's disease. CNS Neurol Disord Drug Targets 2006; 5: 375–380.
Hayashi H, Kimura N, Yamaguchi H, Hasegawa K, Yokoseki T, Shibata M et al. A seed for Alzheimer amyloid in the brain. J Neurosci 2004; 24: 4894–4902.
Kracun I, Rosner H, Drnovsek V, Heffer-Lauc M, Cosovic C, Lauc G . Human brain gangliosides in development, aging and disease. Int J Dev Biol 1991; 35: 289–295.
Brooksbank BW, McGovern J . Gangliosides in the brain in adult Down's syndrome and Alzheimer's disease. Mol Chem Neuropathol 1989; 11: 143–156.
Matsuzaki K . Physicochemical interactions of amyloid [beta]-peptide with lipid bilayers. Biochim Biophys Acta 2007; 1768: 1935–1942.
Haughey NJ, Bandaru VV, Bae M, Mattson MP . Roles for dysfunctional sphingolipid metabolism in Alzheimer's disease neuropathogenesis. Biochim Biophys Acta 2010; 1801: 878–886.
Gylys KH, Fein JA, Yang F, Miller CA, Cole GM . Increased cholesterol in A[beta]-positive nerve terminals from Alzheimer's disease cortex. Neurobiol Aging 2007; 28: 8–17.
Molander-Melin M, Blennow K, Bogdanovic N, Dellheden B, Mansson JE, Fredman P . Structural membrane alterations in Alzheimer brains found to be associated with regional disease development; increased density of gangliosides GM1 and GM2 and loss of cholesterol in detergent-resistant membrane domains. J Neurochem 2005; 92: 171–182.
Yanagisawa K, Odaka A, Suzuki N, Ihara Y . GM1 ganglioside-bound amyloid beta-protein (A beta): a possible form of preamyloid in Alzheimer's disease. Nat Med 1995; 1: 1062–1066.
Yanagisawa K, McLaurin J, Michikawa M, Chakrabartty A, Ihara Y . Amyloid [beta]-protein (A[beta]) associated with lipid molecules: immunoreactivity distinct from that of soluble A[beta]. FEBS Lett 1997; 420: 43–46.
Yanagisawa K, Ihara Y . GM1 ganglioside-bound amyloid beta-protein in Alzheimer's disease brain. Neurobiol Aging 1998; 19: S65–S67.
Yanagisawa K . Role of gangliosides in Alzheimer's disease. Biochim Biophys Acta 2007; 1768: 1943–1951.
Yamamoto N, Hirabayashi Y, Amari M, Yamaguchi H, Romanov G, Van Nostrand WE et al. Assembly of hereditary amyloid beta-protein variants in the presence of favorable gangliosides. FEBS Lett 2005; 579: 2185–2190.
Selkoe DJ . Alzheimer's amyloid of another flavour. Nat Med 1995; 1: 998–999.
Yamamoto N, Van Nostrand WE, Yanagisawa K . Further evidence of local ganglioside-dependent amyloid beta-protein assembly in brain. Neuroreport 2006; 17: 1735–1737.
Mahuran DJ . Biochemical consequences of mutations causing the GM2 gangliosidoses. Biochim Biophys Acta 1999; 1455: 105–138.
Keilani S, Lun Y, Stevens AC, Williams HN, Sjoberg ER, Khanna R et al. Lysosomal dysfunction in a mouse model of Sandhoff disease leads to accumulation of ganglioside-bound amyloid-beta peptide. J Neurosci 2012; 32: 5223–5236.
Tamboli IY, Hampel H, Tien NT, Tolksdorf K, Breiden B, Mathews PM et al. Sphingolipid storage affects autophagic metabolism of the amyloid precursor protein and promotes Abeta generation. J Neurosci 2011; 31: 1837–1849.
Gandy S, Simon AJ, Steele JW, Lublin AL, Lah JJ, Walker LC et al. Days to criterion as an indicator of toxicity associated with human Alzheimer amyloid-beta oligomers. Ann Neurol 2010; 68: 220–230.
Boyd RE, Lee G, Rybczynski P, Benjamin ER, Khanna R, Wustman BA et al. Pharmacological chaperones as therapeutics for lysosomal storage diseases. J Medi Chem 2013; 56: 2705–2725.
Tropak MB, Reid SP, Guiral M, Withers SG, Mahuran D . Pharmacological enhancement of beta-hexosaminidase activity in fibroblasts from adult Tay-Sachs and Sandhoff Patients. J Biol Chem 2004; 279: 13478–13487.
Wustman BA, Boudes P, Lockhart DJ . Stabilizing mutant proteins with small molecules: an example from Fabry Disease. Pediatr Pulm 2012; 47: 114–116.
Osher E, Fattal-Valevski A, Sagie L, Urshanski N, Amir-Levi Y, Katzburg S et al. Pyrimethamine increases beta-hexosaminidase A activity in patients with Late Onset Tay Sachs. Mol Genet Metab 2011; 102: 356–363.
Clarke JT, Mahuran DJ, Sathe S, Kolodny EH, Rigat BA, Raiman JA et al. An open-label Phase I/II clinical trial of pyrimethamine for the treatment of patients affected with chronic GM2 gangliosidosis (Tay-Sachs or Sandhoff variants). Mol Genet Metab 2011; 102: 6–12.
Amicus Therapeutics Announces Positive 12- and 24-Month Data from Phase 3 Fabry Monotherapy Study 011 (Press release). Amicus Therapeutics 2014.
Maegawa GH, Tropak M, Buttner J, Stockley T, Kok F, Clarke JT et al. Pyrimethamine as a potential pharmacological chaperone for late-onset forms of GM2 gangliosidosis. J Biol Chem 2007; 282: 9150–9161.
Mark BL, Mahuran DJ, Cherney MM, Zhao D, Knapp S, James MN . Crystal structure of human beta-hexosaminidase B: understanding the molecular basis of Sandhoff and Tay-Sachs disease. J Mol Biol 2003; 327: 1093–1109.
Bateman KS, Cherney MM, Mahuran DJ, Tropak M, James MN . Crystal structure of beta-hexosaminidase B in complex with pyrimethamine, a potential pharmacological chaperone. J Med Chem 2011; 54: 1421–1429.
Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR . Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000; 404: 770–774.
Petaja-Repo UE, Hogue M, Laperriere A, Bhalla S, Walker P, Bouvier M . Newly synthesized human delta opioid receptors retained in the endoplasmic reticulum are retrotranslocated to the cytosol, deglycosylated, ubiquitinated, and degraded by the proteasome. J Biol Chem 2001; 276: 4416–4423.
Steet RA, Chung S, Wustman B, Powe A, Do H, Kornfeld SA . The iminosugar isofagomine increases the activity of N370S mutant acid beta-glucosidase in Gaucher fibroblasts by several mechanisms. Proc Natl Acad Sci USA 2006; 103: 13813–13818.
Bernier V, Bichet DG, Bouvier M . Pharmacological chaperone action on G-protein-coupled receptors. Curr Opin Pharmacol 2004; 4: 528–533.
Chang EH, Huerta PT . Neurophysiological correlates of object recognition in the dorsal subiculum. Front Behav Neurosci 2012; 6: 46.
Utsumi M, Yamaguchi Y, Sasakawa H, Yamamoto N, Yanagisawa K, Kato K . Up-and-down topological mode of amyloid beta-peptide lying on hydrophilic/hydrophobic interface of ganglioside clusters. Glycoconj J 2009; 26: 999–1006.
Yagi-Utsumi M, Matsuo K, Yanagisawa K, Gekko K, Kato K . Spectroscopic characterization of intermolecular interaction of amyloid beta promoted on GM1 micelles. Int J Alzheimer's Dis 2010; 2011: 925073.
Kakio A, Nishimoto SI, Yanagisawa K, Kozutsumi Y, Matsuzaki K . Cholesterol-dependent formation of GM1 ganglioside-bound amyloid beta-protein, an endogenous seed for Alzheimer amyloid. J Biol Chem 2001; 276: 24985–24990.
Manna M, Mukhopadhyay C . Binding, conformational transition and dimerization of amyloid-beta peptide on GM1-containing ternary membrane: insights from molecular dynamics simulation. PLoS One 2013; 8: e71308.
Fantini J, Yahi N, Garmy N . Cholesterol accelerates the binding of Alzheimer's beta-amyloid peptide to ganglioside GM1 through a universal hydrogen-bond-dependent sterol tuning of glycolipid conformation. Front Physiol 2013; 4: 120.
Hong S, Ostaszewski BL, Yang T, O'Malley TT, Jin M, Yanagisawa K et al. Soluble Abeta oligomers are rapidly sequestered from brain ISF in vivo and bind GM1 ganglioside on cellular membranes. Neuron 2014; 82: 308–319.
Ariga T, Kobayashi K, Hasegawa A, Kiso M, Ishida H, Miyatake T . Characterization of high-affinity binding between gangliosides and amyloid beta-protein. Arch Biochem Biophys 2001; 388: 225–230.
Yamamoto N, Yokoseki T, Shibata M, Yamaguchi H, Yanagisawa K . Suppression of A[beta] deposition in brain by peripheral administration of Fab fragments of anti-seed antibody. Biochem Biophys Res Commun 2005; 335: 45–47.
Acknowledgements
This work was supported by Alzheimer’s Drug Discovery Foundation grant 291003. SHK is supported by BrightFocus Foundation. The β-hex targeted pharmacological chaperone OT1001 was supplied by Amicus Therapeutics. We thank Drs Richie Khanna, Anadina Garcia and Michelle Frascella for their assistance in animal handling and dosing, and Drs Elfrida Benjamin and Rick Hamler for their assistance in measuring OT1001 levels in the PK and PD studies conducted in C57BL/J6 mice.
Author Contributions
EMK, HNW, ACS, SHK, REB, DJL and ERS designed the experiments with MEE BAW and SG. EMK, HNW, ACS, SHK, ADM, JCK, REB and ERS performed the experiments. EMK, HNW, ACS and SHK analyzed the data. EMK, BAW and SG wrote the manuscript. WLK and KY provided material support. JWS and KY critically read the manuscript for intellectual content. MEE, BAW and SG supervised the project.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Within the past 5 years, SG has held research grants from Baxter Pharmaceuticals and Amicus Therapeutics. SG is also a member of the Data and Safety Monitoring Board for the Pfizer-Janssen Alzheimer's Immunotherapy Alliance. BAW, ACS, DJL, JWS and ERS are employees or shareholders of OrPhi Therapeutics. HNW and REB are employees and shareholders of Amicus Therapeutics.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Knight, E., Williams, H., Stevens, A. et al. Evidence that small molecule enhancement of β-hexosaminidase activity corrects the behavioral phenotype in Dutch APPE693Q mice through reduction of ganglioside-bound Aβ. Mol Psychiatry 20, 109–117 (2015). https://doi.org/10.1038/mp.2014.135
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2014.135
This article is cited by
-
Insulin resistance: a connecting link between Alzheimer’s disease and metabolic disorder
Metabolic Brain Disease (2021)
-
Mouse models of neurodegenerative disease: preclinical imaging and neurovascular component
Brain Imaging and Behavior (2018)
-
Lysosomal dysfunction in the brain of a mouse model with intraneuronal accumulation of carboxyl terminal fragments of the amyloid precursor protein
Molecular Psychiatry (2017)
-
Unexpected partial correction of metabolic and behavioral phenotypes of Alzheimer’s APP/PSEN1 mice by gene targeting of diabetes/Alzheimer’s-related Sorcs1
Acta Neuropathologica Communications (2016)
-
Pharmacological Chaperone Therapy: Preclinical Development, Clinical Translation, and Prospects for the Treatment of Lysosomal Storage Disorders
Molecular Therapy (2015)
