
Abstract
Gastrointestinal stromal tumors (GISTs) are mesenchymal tumors usually driven by the mutational activation of receptor tyrosine kinases, KIT, or PDGFRA. Oncogenic activation of phosphatidylinositide-3-kinase (PI3K), a downstream effector in the KIT signaling pathway, has been identified in different types of cancer, with the PI3K 110α subunit encoded by PIK3CA being a common mutational target. In this study, the mutational hotspot in the PIK3CA kinase domain encoded by exon 20 was evaluated in 529 imatinib-naive GISTs using PCR amplification and Sanger sequencing. Eight mutations (two co-existing in one tumor) were identified. Subsequently, The cobas PIK3CA Mutation Test was employed to evaluate mutational hotspots in exons 1, 4, 7, and 9 in 119 PIK3CA exon 20-wild type tumors. In two cases, mutations in exons 1 and 9 were identified. In one GIST, previously undetected by Sanger sequencing, the exon 20 mutation was discovered. Altogether, eight primary and two metastatic GISTs carried PIK3CA mutations. The size of primary PIK3CA-mutant GISTs was ≥14 cm (mean size 17 cm), and mitotic activity varied from 0 to 72 per 50HPF (mean 5/50HPF). Follow-up data showed short survival in 6 of 7 studied cases. Detection of PIK3CA mutations in large or metastatic KIT-mutant GISTs may suggest that PIK3CA-mutant clones have a proliferative advantage during disease progression. Tyrosine kinase inhibitors have been successfully used in GIST treatment. However, resistance frequently develops due to secondary KIT mutations or activation of downstream to KIT signaling pathways, such as the PI3K/AKT/mTOR pathway. PIK3CA mutations similar to the ones detected in GISTs have been shown to cause such activation. Therefore, genotyping of PIK3CA in GISTs might help to pinpoint primary and metastatic tumors with the potential to develop resistance to tyrosine kinase inhibitors and guide therapy with PI3K inhibitors.
Similar content being viewed by others


Main
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the gastrointestinal (GI) tract. In general, GISTs are driven by mutational, oncogenic activation of receptor tyrosine kinases, KIT, or platelet-derived growth factor receptor α (PDGFRA), although subsets of KIT/PDGFRA-wild-type tumors have been identified and include succinate dehydrogenase (SDH)-deficient GISTs, neurofibromatosis 1 (NF1)-associated GISTs, BRAF mutant GISTs, and the so-called ‘quadruple wild-type tumors’ that lack mutations in KIT, PDGFRA, BRAF, and SDH genes.1, 2 Phosphatidylinositol 3-kinase, catalytic, alpha (PIK3CA) encodes the p110α catalytic subunit of phosphatidylinositide-3-kinase (PI3K), a lipid kinase, downstream effector in the KIT signaling pathway that promotes cellular growth, proliferation, and survival. Activation of PI3K/AKT/mTOR signaling pathway has been reported in different types of human cancer, and is often caused by PIK3CA mutations.3 PIK3CA mutations frequently coexist with mutational activation of other oncogenes, such as BRAF and RAS.4 Because the presence of PIK3CA mutations might negate the effect of the KIT inhibitor and indicate tumor response to PI3K/AKT/mTOR axis inhibitors, the mutation status of PIK3CA could be a factor in the selection of targeted therapy.5, 6 Recently, PIK3CA mutations have been reported in two primary gastric GISTs: one concurrent with a KIT mutation and another with the HRAS mutation, and in treatment-resistant metastasis of a BRAF-mutant GIST.7, 8, 9 Thus, activation of the PI3K/AKT/mTOR signaling pathway may have a role in both pathogenesis and the progression of GISTs. The presence of the PIK3CA mutation could indicate an alternative treatment inhibiting the PI3K/AKT/mTOR signaling pathway for patients with advanced and metastatic tumors, as opposed to KIT inhibitor therapy. The aim of this study was to determine the frequency of PIK3CA mutations in imatinib-naive GISTs and their role in GIST biology and pathogenesis.
Materials and methods
Study Material
The analyzed cohort of 529 GISTs contained primary tumors from different locations, including the esophagus (n=7), stomach (n=214), small intestine (n=127), large intestine with rectum and anus (n=32), and intra-abdominal (n=141) and metastatic extra-abdominal GISTs (n=4). The intra-abdominal category included tumors involving omentum, retroperitoneum, and GISTs largely disseminated in the abdominal cavity at the time of diagnosis. In all cases, GIST diagnosis was confirmed by positive KIT (CD117) and/or ANO1 (DOG1) immunohistochemistry. Among gastric GISTs, 16 SDH-deficient tumors were identified following SDHB and SDHA immunohistochemistry. In the majority of cases, CD34, SMA, and S100 immunohistochemistry and KIT, PDGFRA, BRAF, HRAS, and KRAS genotyping data were available. Tumor size was documented in 347 primary GISTs. There were 195 tumors <10 cm and 152 tumors >10 cm. Furthermore, in 71 cases, intra-abdominal dissemination or extra-abdominal metastases were present at the diagnosis.
Study Design
A total of 529 GISTs were examined for PIK3CA exon 20 mutations by PCR amplification and Sanger sequencing. In addition, 119 cases were examined by The cobas PIK3CA Mutation Test (Roche Molecular Systems, Pleasanton, CA, USA) in order to detect mutations in other exons, as their detection by Sanger sequencing is hampered by homologous pseudogene sequences. Seventy-five GISTs evaluated by The cobas PIK3CA Mutation Test were either large (>10 cm) or metastatic tumors. In addition, two cases were analyzed by the Ion Torrent next generation sequencing platform (Life Technologies/Thermo Fisher Scientific, Waltham, MA, USA) to further confirm Sanger sequencing results. In one case, a double PIK3CA mutation was further analyzed through cloning and sequencing.
Molecular Studies
DNA extraction
DNA was extracted from formalin-fixed paraffin-embedded tissue samples. Five to ten 5-μ-thick sections were deparaffinized with xylene, washed twice in ethanol, lyophilized, and incubated with 10 μg/μl proteinase K (Roche Diagnostics, Indianapolis, IN, USA) in Hirt-Buffer at 55 °C for 24 h. Subsequently, DNA was recovered using the Maxwell 16 robotic system and DNA IQ Casework Pro Kit (Promega, Madison, WI, USA).
PCR amplification and Sanger sequencing
PIK3CA exon 20 was PCR amplified with AmpliTaq Gold DNA polymerase (Applied Biosystems by Life Technologies, Austin, TX, USA) following a standard three-temperature protocol with denaturing at 94 °C, annealing at 48 °C, and extension at 72 °C. Two sets of primers were used: (1) PIK3CA1F 5′-AGGAGATGTGTTACAAGGCT-3′ and PIK3CA1R 5′–TTGTGTGGAAGATCCAATCC-3′ and (2) PIK3CA2F 5′-TGCATACATTCGAAAGACCC-3′ and PIK3CA2R 5′-TTGTGTGGAAGATCCAATCC-3′. The PCR product sizes for these two assays were 243 and 128 bp, respectively. PCR amplification products were purified using the QIAquick Gel Extraction Kit (Qiagen, Valencia, CA, USA) following agarose gel electrophoresis and then sequenced directly with forward and reverse primers. Cases revealing sequence variations were evaluated at least twice to ensure reproducibility of the PCR amplification and sequencing results. In one case, PIK3CA exon 20 PCR amplification products were cloned into plasmid using the TOPO TA Cloning Kit (Invitrogen by Life Technologies) and subsequently sequenced. The sequences were analyzed following alignment with PIK3CA, NG_012113.2 and NM_006218.2:c reference sequences (www.ncbi.nml.nih.gov). Known PIK3CA mutant colon carcinomas were used as a positive controls. The Sanger sequencing service was performed by MacrogenUSA (Rockville, MD, USA).
The cobas PIK3CA Mutation Test
The cobas PIK3CA Mutation Test (Roche Molecular Systems, Pleasanton, CA, USA), a real-time PCR assay, allows detection of mutations affecting the following codons: p.R88 in exon 1, p.N345 in exon 4, p.C420 in exon 7, p.E542, p.E545 and p.Q546 in exon 9, and p.M1043, p.H1047, and p.G1049 in exon 20. Genotyping using The cobas PIK3CA Mutation Test was performed following the protocol provided by the manufacturer.
Next-generation sequencing
Next-generation sequencing was performed using the Ion Torrent NGS platform in accordance with the manufacturer’s instructions (Life Technologies/Thermo Fisher Scientific, Waltham, MA, USA). Libraries were prepared using 10 ng of tumor DNA, Ion AmpliSeq Library Kit 2.0, Ion AmpliSeq Cancer Hotspot Panel v2 Kit, and Ion Xpress Barcode Adapters 1-16 Kit. Template preparation was done using emulsion PCR with Ion OneTouch 200 Template Kit v2 DL and Ion OneTouch DL and Ion OneTouch ES devices. Sequencing was performed using a Personal Genome Machine (PGM) and the Ion PGM 200 Sequencing Kit. The data were processed by Torrent Server Suite 4.2 and sequences aligned to the human genome reference sequence HG-19 (The Genome Reference Consortium). Variant calling was performed using Variant Caller v4.2, which is compatible with the Integrative Genomics Viewer (Broad Institute, Cambridge, MA, USA), a high-performance visualization tool for interactive exploration of large, integrated data sets.
Results
Molecular Studies
Sanger sequencing
Sanger sequencing of the PCR amplified PIK3CA exon 20 revealed eight mutations in seven GISTs. Examples of Sanger sequencing are shown in Figure 1. All mutations consisted of single-nucleotide substitutions: c.3103G>A, c.3127A>G, c.3137C>T, c.3139C>T, c.3140A>G, c.3140A>T, c.3143A>G, and c.3158C>T. At the protein level, these substitutions would lead to p.A1035T, p.M1043V, p.A1046V, p.H1047R, p.H1047L, p.H1047Y, p.H1048R, and p.T1053I mutations, respectively. In one tumor, two substitutions, c.3103G>A and c.3158C>T, were detected. The cloning and sequencing of PCR products confirmed the presence of these mutations on two different alleles. All mutations identified in this study were previously reported in other tumors (Catalog of somatic mutations in cancer, cancer.sanger.ac.uk/cosmic).

Examples of PIK3CA mutations detected by at least two independent PCRs and Sanger sequencing.
In three cases, initially identified substitutions (c.3117C>T, c.3152G>A, and c.3153G>A, the cause of p.F1039F and p.W1051* PIK3CA mutations) could not be confirmed by the sequencing of two independent PCR amplification products (Figure 2). Subsequently, two of these cases were evaluated by Ion Torrent next-generation sequencing, which did not confirm the presence of clones with c.3152G>A and c.3153G>A PIK3CA mutations. However, due to the poor DNA quality, the sequencing coverage of the analyzed regions was only 353 and 145 reads, respectively.

Two PIK3CA mutations initially detected by PCR amplification and Sanger sequencing that could not be confirmed by independent assays.
The cobas PIK3CA Mutation Test
Three additional PIK3CA mutants were identified in 120 GISTs evaluated using The cobas PIK3CA Mutation Test (Table 1). These mutants involved p.R88 in exon 1, p.E545 in exon 9, and p.H1047 in exon 20. The latter mutant was not detected via Sanger sequencing, while the two former ones were not tested by that assay.
Clinicopathological Profile of PIK3CA Mutant GISTs
The clinicopathologic data of PIK3CA mutants are summarized in Table 2. The 10 patients included 7 men and 3 women (mean age, 58 years). There were four gastric and one small intestinal primary GIST, one liver metastasis of a small bowel GIST, three retroperitoneal GISTs with small intestinal GIST-like morphology, and one GIST metastatic to the abdominal wall from an unknown primary tumor. The tumor size of all primary PIK3CA mutant GISTs was ≥14 cm (mean and median size, 17 cm), and mitotic activity varied from 0 to 72 per 50HPF (median, 5/50 HPFs).
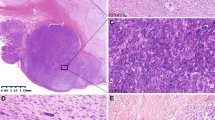
Nine PIK3CA mutant GISTs showed spindle cell or spindle to ovoid cell morphology (Figure 3). Epithelioid cell morphology was seen in the abdominal wall metastatic GIST with unknown primary tumor. All tumors were KIT and DOG1 positive, while CD34 and SMA were expressed in seven and one cases, respectively (Figure 4). The follow-up available in the seven cases revealed death of the disease for two cases, with median survival time <9 months and overall short survival <35 months in four additional cases (in which the cause of death was not available). PIK3CA mutations were concurrent with KIT exon 11 mutations in all 9 analyzed cases.

Four examples of PIK3CA mutant GISTs. (a) An epithelioid GIST of the stomach. (b) A retroperitoneal spindle cell GIST with Verocay body-like, prominent aggregates of cell processes, typically seen in GISTs of intestinal origin. (c) A GIST with spindled to ovoid cell morphology. (d) A spindle cell GIST representing the morphology often seen in intestinal GISTs.

Immunohistochemical findings in PIK3CA mutant GISTs. The tumor cells are positive for KIT, DOG1, CD34, and SMA.
Frequency of PIK3CA Mutations in GIST
PIK3CA exon 20 mutations were identified by Sanger sequencing in 1.3% (7 of 529) of analyzed GISTs. However, if tumors <10 cm were excluded, then the frequency rose up to 2.7% (6 of 223). In a similar cohort, the frequency of PIK3CA mutations other than those in the exon 20 PIK3CA hot-spot was 2.7% (2 of 75), as documented by The cobas PIK3CA Mutation Test. Also, this real-time PCR assay was more sensitive than Sanger sequencing and identified one additional exon 20 mutant previously undetected by the Sanger sequencing. Based on the above data, we estimated that PIK3CA mutations in large (>10 cm) and advanced GISTs might reach a 5–6% frequency.
Discussion
Phosphatidiylinositol 3-Kinase, or PI3K, is a lipid kinase, a downstream effector in the KIT signaling pathway that is thought to promote processes, such as cellular growth, proliferation, and survival. Oncogenic activation of the PI3K pathway has been implicated in different types of cancer. Activation of the PI3K/AKT/mTOR pathway contributes to a more aggressive tumor phenotype, which results in shorter survival and poor treatment outcome.10 Genetic or epigenetic mutations affecting PI3K, PI3K downstream kinases, such as AKT or mammalian target of rapamycin (mTOR), and PTEN, a major PI3K-negative regulator, have been reported.3, 5, 6
Relatively little is known about the genetic alteration of PI3K/AKT/mTOR pathway in GISTs, although one study reported frequent mono-allelic loss of PTEN, a negative regulator of PI3K.11, 12 In addition, activation of the PI3K/AKT pathway was proposed to be involved in tumor cell survival in imatinib-sensitive and -resistant GISTs.13
PIK3CA, a gene that encodes the p110α catalytic subunit of PI3K, has been shown to be mutated in common carcinomas, malignant melanoma, and some sarcomas. The majority of PIK3CA mutations cluster in exon 9 and exon 20, encoding helical and kinase domains respectively, although activating mutations in other PIK3CA regions including exons 1, 4, and 7 have been reported.3
In this study, a large cohort of imatinib-naive GISTs was screened for PIK3CA mutations and 10 mutants were identified. A great majority of these mutations were located in exon 20, with four of them affecting codon 1047. This codon was mutated in all three previously reported PIK3CA mutant GISTs.7, 8, 9 Thus, similarly to other cancer, p.H1047 appears to be the most common mutational hotspot in GISTs. One tumor revealed two co-existing PIK3CA exon 20 mutations. The co-existence of two different PIK3CA mutations has been previously reported in breast cancer and linked to more aggressive clinical outcomes.14, 15
A previous study based on immunohistochemistry suggested that activation of the mTOR signaling pathway is characteristic for the PDGFRA mutant and wild-type GISTs, rather than KIT mutant GISTs.16 In our study, all nine analyzed PIK3CA mutants turned out to be KIT exon 11 mutants. Similarly, one of three previously reported PIK3CA mutant GISTs carried KIT K558_E562del, typical of GISTs.7
One PIK3CA mutation was reported in KIT/PDGFRA-wild type, BRAF V600E mutant GIST metastasis during BRAF-inhibitor treatment, at the time of disease progression.8 Primary GISTs driven by BRAF mutations are exceptionally rare.7, 17 In the current study, PIK3CA was evaluated in one BRAF mutant GIST and revealed wild-type sequences.
Nevertheless, a p.H1047R PIK3CA mutation co-existing with a p.G12V HRAS mutation was identified by next-generation sequencing in a high-grade gastric tumor reported as ‘quadruple wild type’ GIST.9 Thus, in rare cases, PIK3CA mutations might have a role in wild-type GIST pathogenesis. In our study, no tumors with co-existing PIK3CA and HRAS or KRAS mutations were identified by Sanger sequencing.
Previous studies documented p.G12R and p.G12V KRAS mutations, respectively, in an imatinib-resistant lesion of disseminated GIST and in an anaplastic KIT-negative tumor developed following long-term imatinib therapy for CML.9, 18 Based on previous studies, the frequency of RAS mutations in imatinib-naive GISTs remains very low.19 More recently, a KRAS G12V mutation at 29% allele frequency has been reported in an SDH-deficient highly malignant gastric GIST.20 No PIK3CA, HRAS, and KRAS mutations were identified in 16 SDH-deficient GISTs analyzed in this study.
In this study, all GISTs harboring PIK3CA mutations were large (>10 cm) tumors. Also, previously published PIK3CA mutants represented high-grade primary or metastatic GISTs. The presence of PIK3CA mutations in such tumors may suggest that PIK3CA mutant clones have proliferative advantage and can become dominant in a late stage of the GIST genetic evolution. These mutations can also arise without the selection pressure of a KIT tyrosine kinase inhibitor, such as imatinib mesylate.
Imatinib mesylate, a KIT and PDGFRA tyrosine kinase inhibitor, has been used for the treatment of primary advanced and disseminated GISTs. However, both primary and secondary resistance acquired after or during initially successful treatment occurs. 21 The resistance could be related to the mutational activation of downstream pathways, such as the PI3K/AKT/mTOR pathway. Thus, identification of PIK3CA mutations in primary KIT mutant GIST may have significant clinical implications in the choice of a kinase inhibitor therapy, as the mutational activation of PI3K may nullify the effect of KIT inhibition due to independent downstream activation of the signaling pathways. Also, the type of PIK3CA mutation might influence the response to PI3K/AKT/mTOR inhibitors.22
Applicability of Sanger sequencing in the analysis of PIK3CA mutations has limitations. The presence of the pseudogene on chromosome 22 with high (>95%) homology to the PIK3CA exon 9 sequence hampers detection of mutations by Sanger sequencing. Thus, in this study, The cobas PIK3CA Mutation Test was employed to search for common PIK3CA exon 9 mutations. This real-time PCR assay, developed to detect changes in PIK3CA mutational hotspots, is more sensitive than Sanger sequencing.23 In our study, the comparison of these two methods revealed a high concordance with only two variances. In one case, a rare mutation identified by Sanger sequencing was not detectable by The cobas PIK3CA Mutation Test and, in the second case, Sanger sequencing turned to be less sensitive in the detection of a mutant minority clone.
The reproducibility of Sanger sequencing results was generally good. However, in three cases, initially detected heterozygous G>A and C>T mutations were not seen in subsequent PCR amplifications. Although such results might be a consequence of intratumor heterogeneity,24 the next-generation sequencing failed to detect subclones carrying these mutations. Because the C:G>T:A substitutions represent the most common sequencing artifacts detected in PCR of formalin-fixed paraffin-embedded DNA,25 it is possible that early sequencing results from these two cases represented random PCR amplification artifacts. Studies of multiple, well-preserved DNA samples with next-generation ultra-deep sequencing could help to distinguish intratumor clonal heterogeneity from random PCR amplification artifacts.26
In summary, we reported the rare occurrence of PIK3CA mutations in imatinib-naive GISTs. These mutations occurred predominantly in large and clinically aggressive GISTs, suggesting that the expansion of PIK3CA mutant clones is an additional oncogenic event that can also develop without selection pressure caused by a kinase inhibitor. Detection of these mutations is significant for the selection of targeted therapy as their presence may preclude the use of a tyrosine kinase inhibitor and instead require inhibitors for the PI3K/AKT/mTOR pathway.
References
Miettinen M, Lasota J . Gastrointestinal stromal tumors. Gastroenterol Clin North Am 2013;42:399–415.
Pantaleo MA, Nannini M, Corless CL et al. Quadruple wild-type (WT) GIST: defining the subset of GIST that lacks abnormalities of KIT, PDGFRA, SDH, or RAS signaling pathways. Cancer Med 2015;4:101–103.
Samuels Y, Waldman T . Oncogenic mutations of PIK3CA in human cancers. Curr Top Microbiol Immunol 2010;347:21–41.
Janku F, Lee JJ, Tsimberidou AM et al. PIK3CA mutations frequently coexist with RAS and BRAF mutations in patients with advanced cancers. PLoS One 2011;6:e22769.
Fruman DA, Rommel C . PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 2014;13:140–156.
Thorpe LM, Yuzugullu H, Zhao JJ . PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer 2015;15:7–24.
Daniels M, Lurkin I, Pauli R et al. Spectrum of KIT/PDGFRA/BRAF mutations and phosphatidylinositol-3-kinase pathway gene alterations in gastrointestinal stromal tumors (GIST). Cancer Lett 2011;312:43–54.
Falchook GS, Trent JC, Heinrich MC et al. BRAF mutant gastrointestinal stromal tumor: first report of regression with BRAF inhibitor dabrafenib (GSK2118436) and whole exomic sequencing for analysis of acquired resistance. Oncotarget 2013;4:310–315.
Serrano C, Wang Y, Mariño-Enríquez A et al. KRAS and KIT gatekeeper mutations confer polyclonal primary imatinib resistance in GI stromal tumors: relevance of concomitant phosphatidylinositol 3-kinase/AKT dysregulation. J Clin Oncol 2015;33:e93–e96.
Liao X, Morikawa T, Lochhead P et al. Prognostic role of PIK3CA mutation in colorectal cancer: cohort study and literature review. Clin Cancer Res 2012;18:2257–2268.
Quattrone A, Wozniak A, Dewaele B et al. Frequent mono-allelic loss associated with deficient PTEN expression in imatinib-resistant gastrointestinal stromal tumors. Mod Pathol 2014;27:1510–1520.
Patel S . Exploring novel therapeutic targets in GISTs: focus on the PI3K/Akt/mTOR pathway. Curr Oncol Rep 2013;15:386–395.
Floris G, Wozniak A, Sciot R et al. A potent combination of the novel PI3K inhibitor, GDC-0941, with imatinib in gastrointestinal stromal tumor xenografts: long-lasting responses after treatment withdrawal. Clin Cancer Res 2013;19:620–630.
Stemke-Hale K, Gonzalez-Angulo AM, Lluch A et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res 2008;68:6084–6091.
Cizkova M, Susini A, Vacher S et al. PIK3CA mutation impact on survival in breast cancer patients and in ERa, PR and ERBB2-based subgroups. Breast Cancer Res 2012;14:R28.
Sápi Z, Füle T, Hajdu M et al. The activated targets of mTOR signaling pathway are characteristic for PDGFRA mutant and wild-type rather than KIT mutant GISTs. Diagn Mol Pathol 2011;20:22–33.
Agaram NP, Wong GC, Guo T et al. Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer 2008;47:853–859.
Antonescu CR, Romeo S, Zhang L et al. Dedifferentiation in gastrointestinal stromal tumor to an anaplastic KIT-negative phenotype: a diagnostic pitfall: morphologic and molecular characterization of 8 cases occurring either de novo or after imatinib therapy. Am J Surg Pathol 2013;37:385–392.
Lasota J, Xi L, Coates T et al. No KRAS mutations found in gastrointestinal stromal tumors (GISTs): molecular genetic study of 514 cases. Mod Pathol 2013;26:1488–1491.
Hechtman JF, Zehir A, Mitchell T et al. Novel oncogene and tumor suppressor mutations in KIT and PDGFRA wild type gastrointestinal stromal tumors revealed by next generation sequencing. Genes Chromosomes Cancer 2015;54:177–184.
Liegl B, Kepten I, Le C et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol 2008;216:64–74.
Janku F, Wheler JJ, Naing A et al. PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR signaling pathway inhibitors in early-phase clinical trials. Cancer Res 2013;73:276–284.
Schlag P, Hoeppner C, Bristol A et al. A real-time PCR assay for the detection of PIK3CA mutations in formalin-fixed paraffin embedded tissue (FFPET) specimens of breast cancer (BC). Cancer Res 2013;73:4217.
Swanton C . Intratumor heterogeneity: evolution through space and time. Cancer Res 2012;72:4875–4882.
Do H, Dobrovic A . Sequence artifacts in DNA from formalin-fixed tissues: causes and strategies for minimization. Clin Chem 2015;61:64–71.
Stenzinger A, Endris V, Pfarr N et al. Targeted ultra-deep sequencing reveals recurrent and mutually exclusive mutations of cancer genes in blastic plasmacytoid dendritic cell neoplasm. Oncotarget 2014;5:6404–6413.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Lasota, J., Felisiak-Golabek, A., Wasag, B. et al. Frequency and clinicopathologic profile of PIK3CA mutant GISTs: molecular genetic study of 529 cases. Mod Pathol 29, 275–282 (2016). https://doi.org/10.1038/modpathol.2015.160
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2015.160
This article is cited by
-
New treatment strategies for advanced-stage gastrointestinal stromal tumours
Nature Reviews Clinical Oncology (2022)
-
Prevalence and significance of M541L single nucleotide polymorphism in the central European cohort of gastrointestinal stromal tumor patients
Journal of Cancer Research and Clinical Oncology (2021)
