
Abstract
Parosteal osteosarcoma, low-grade central osteosarcoma, and fibrous dysplasia share similar histological features that may pose a diagnostic challenge. The detection of GNAS mutations in primary bone tumors has been useful in clinical practice for diagnosing fibrous dysplasia. However, the recent report of GNAS mutations being detected in a significant proportion of parosteal osteosarcoma challenges the specificity of this mutation. As the number of cases reported in this study was small we set out to determine if these results could be reproduced. We studied 97 formalin-fixed paraffin-embedded low-grade osteosarcomas from 90 patients including 62 parosteal osteosarcomas, of which MDM2 amplification was detected in 79%, 11 periosteal osteosarcomas and 24 low-grade central osteosarcoma samples. The mutational status of GNAS was analyzed in codons p.R201, p.Q227, and other less common GNAS alterations by bidirectional Sanger sequencing and/or next generation sequencing using the Life Technologies Ion Torrent platform. GNAS mutations were not detected in any of the low-grade osteosarcomas from which informative DNA was extracted. Our findings therefore support prior observations that GNAS mutations are highly specific for fibrous dysplasia and occur rarely, if ever, in parosteal and other low-grade osteosarcomas.
Similar content being viewed by others

Main
Parosteal and low-grade central osteosarcomas are rare low-grade primary malignant bone tumors comprising <4% of all osteosarcomas. In contrast, fibrous dysplasia is one of the most common benign fibro-osseous tumor-like lesions in medullary bone. Morphologically, there is considerable overlap between these lesions all showing variably shaped bony trabeculae surrounded by spindled-shaped cells, with little to no cytologic atypia.1 In the majority of cases, imaging studies can easily differentiate parosteal osteosarcoma from fibrous dysplasia and central low-grade osteosarcoma: parosteal osteosarcoma is a surface tumor that may secondarily invade bone marrow, and low-grade central osteosarcoma and fibrous dysplasia are centrally based. Occasionally, fibrous dysplasia arises eccentrically in the marrow space, presenting as an exophytic surface-based lesion. Such lesions are referred to as fibrous dysplasia protuberans.2 Hence, distinguishing fibrous dysplasia protuberans from parosteal osteosarcoma can be challenging and differentiating between low-grade central osteosarcoma and fibrous dysplasia can also be difficult.
Parosteal osteosarcoma typically harbors one or more supernumerary ring chromosomes with amplification of the MDM2, SAS, and CDK4 genes.3, 4 MDM2 amplification is reported to occur in 93% (14/15) of low-grade central osteosarcomas3, 5 and in 79% (68/86) of parosteal osteosarcomas.5, 6 Dedifferentiated parosteal osteosarcoma is a distinct tumor variant in which a (high-grade) sarcoma coexists with a conventional parosteal osteosarcoma. Dedifferentiation affects 16–43% of parosteal osteosarcomas with the characteristic gene amplifications being retained and can be present at the first presentation (synchronous type) or at the time of recurrence (metachronous type).7, 8, 9 Bone tumors analyzed previously for GNAS alterations include fibrous dysplasia (N=405),10, 11, 12, 13, 14, 15, 16 ossifying fibroma (N=65),10, 12, 15, 16 osteofibrous dysplasia (N=19),10, 12, 13 low-grade central osteosarcoma (N=12)10, 13, 14, and parosteal osteosarcoma (N=10).10 GNAS mutations have been reported in fibrous dysplasia and have not been described until recently in any other fibrous osseous lesions with the exception of a single low-grade central osteosarcoma.14 These reported GNAS mutations in fibrous dysplasia include p.R201C,13 p.R201H,13 p.R201S,17 and p.Q227L.13 A GNAS mutation in codon 201 (p.R201G) has been found in lesions in McCune–Albright syndrome.18 Other GNAS mutations including p.R201P, p.R201L, p.R201S, p.Q227R, p.Q227H, and p.Q227K have been detected in sporadic endocrine tumors.19, 20 As fibrous dysplasia may be a component of the mosaic disorder in McCune–Albright syndrome, it would not be surprising if any of the above mutations were detected occasionally in fibrous dysplasia. Notably, GNAS mutations have been reported to occur in up to 90% of fibrous dysplasias with p.R201 involving 95% of cases; mutations p.R201H and p.R201C occurring in 57 and 38%, respectively, and p.Q227L being detected in ~5%.13 The GNAS p.K233N has only been detected in one case of colon adenocarcinoma, described in the Catalogue of Somatic Mutations in Cancer (COSMIC) with the mutation ID COSM192557. More recently, GNAS R201 substitutions have been reported in 88% of intraductal papillary mucinous tumors of the pancreas (88%) and in 16 of 32 low-grade appendiceal mucinous neoplasms.21
The mutual exclusiveness of GNAS mutations and MDM2 amplifications has been analyzed only in a small study by Tabareau-Delalande et al.10 who demonstrated the absence of GNAS mutations in 10 parosteal osteosarcomas that harbored MDM2 amplifications.10 It is therefore of particular interest that Carter et al.22 reported that 5/9 parosteal osteosarcomas harbored GNAS mutations. Specifically, they identified GNAS p.R201C mutations in four cases and p.R201H in another case. Of the five parosteal osteosarcomas, two additionally revealed MDM2 amplifications.22 This recent publication questions the reliability of using GNAS alterations for supporting a diagnosis of fibrous dysplasia and excluding a diagnosis of low-grade central and parosteal osteosarcomas. The aim of this study was to extend the study undertaken by Carter et al. to assess the frequency of GNAS alterations in a larger set of low-grade osteosarcomas.
Materials and methods
Patients and Samples
The study included review of the histology and clinical notes from 40 patients with osteosarcoma (29 parosteal, 6 periosteal, and 5 low-grade central) from the files at the Royal National Orthopaedic Hospital, and from 50 patients with osteosarcoma (26 parosteal, 5 periosteal, and 19 low-grade central) from the University Hospital Basel (Table 1). Ethical approval was obtained from the Cambridgeshire 2 Research Ethics Service (reference 09/H0308/165), the University College London Biobank for Health and Disease ethics committee (covered by the Human Tissue Authority license 12055: project EC17.1), and from the Ethikkommission beider Basel (reference 274/1).
DNA Extraction
Representative formalin-fixed paraffin-embedded tissue blocks were selected following review by at least two experienced bone tumor pathologists. DNA was extracted from tumor-rich areas (>60% tumor). Five to 10 consecutive 10-μm thick sections were cut and placed in eppendorf tubes (Eppendorf, Hamburg, Germany). If tumor enrichment was required, unstained sections were microdissected manually using a 14 g hypodermic needle.23 Genomic DNA was extracted from the formalin-fixed paraffin-embedded material using QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany). DNA was extracted from separate blocks containing conventional and dedifferentiated components of parosteal osteosarcoma. DNA concentration and quality were assessed by Nanodrop spectrophotometer (ThermoFisher, Wilmington, DE, USA) and the Qubit dsDNA HS Assay Kit (Life Technologies, Carlsbad, CA, USA).
MDM2 Amplification by Fluorescent In Situ Hybridization
Fluorescent in situ hybridization for MDM2 amplification was performed as previously described on 5-μm sections cut from representative tissue blocks of all cases (Royal National Orthopaedic Hospital and University Hospital Basel).24 The fluorescent in situ hybridization probe for MDM2 on chromosome 12 with its respective chromosome enumeration probe was purchased from Zytovision (Zytovision GmbH, Bremerhaven, Germany), Zytolight SPEC MDM2/CEN12 (centromere 12; Z-2013) Dual Colour Probes. Non-informative cases were excluded from the analysis. The number of each signal for MDM2 (green) and CEN12 (red) was scored by counting a minimum of 50 non-overlapping nuclei per case, and the average number of MDM2 and CEN12 signals was then calculated. A ratio of >2.0 was considered to represent MDM2 amplification, a ratio of ≤2 was considered to be non-amplified. All fluorescent in situ hybridization was reviewed by at least two experienced individuals.
Next Generation Sequencing
Genomic DNA (≥10 ng) was used for multiplex polymerase chain reaction (PCR) amplification (50 amplicons panel). The resulting amplicons were barcoded (Fluidigm, South San Francisco, CA USA) and the DNA libraries were generated using the Fluidigm Access Array System (Fluidigm). Emulsion PCR was performed using the Ion Personal Genome Machine Template OT2 200 prep kit and the One Touch 2 System (Life Technologies). Templates were enriched to at least 70% of template-positive Ion Sphere particles and the enrichment was checked with the Ion Sphere Quality Control Kit (Life Technologies). Sequencing was performed on a 316 Chip v2 (Ion Sequencing 200 kit v2, Life Technologies) and run on the Ion Torrent Personal Genome Machine PGM (Life Technologies). Primers were designed with the Primer3 software (http://bioinfo.ut.ee/primer3-0.4.0/primer3/) for GNAS mutation hotspots including the missense mutations in codons p.R201, p.Q227, and p.K233 (Supplementary Table 1). Reads were aligned to the reference genome hg19 and BAM files were generated using Ion torrent suite version 4.0.1. Reads were visualized using Integrative Genomics Viewer (IGV v 2.2 Broad Institute, Boston, MA, USA) (https://www.broadinstitute.org/igv/) with the appropriate BED files. A mutation was deemed present when there was ≥5% mutation present in a region with ≥500 reads. Six cases of fibrous dysplasia known to harbor GNAS mutations were included as control samples.
Sanger Sequencing
Bidirectional Sanger sequencing for GNAS codons p.R201 in exon 8 and codon p.Q227 in exon 9 was performed for all cases. Each PCR was carried out with 0.4 μM of GNAS p.R201 exon 8 and GNAS p.Q227 exon 9 primers (Supplementary Table 1). The PCR cycling parameters included initial denaturation at 94 °C for 2 min, 40 cycles of denaturation at 94 °C for 30 s, annealing at 62 °C for 30 s, and extension at 72 °C for 1 min, followed by a final extension at 72 °C for 10 min. The PCR products of GNAS p.R201 exon 8 (159 bp) and GNAS p.Q227 exon 9 (174 bp) were separated on a 6% polyacrylamide gel and visualized by ethidium bromide staining. The DNA products were purified with the DNA Clean-up & Concentrator (Zymo Research, Irvine, CA, USA) and aliquots of 30 ng of the PCR products were used for the bidirectional Sanger sequencing (Beckman Coulter). The sequencing chromatograms were analyzed using the FinchTV software (Perkin Elmer, Waltham, MA, USA).
Mutation-Specific Restriction Enzyme Digest for GNAS Mutations
Cases from the Royal National Orthopaedic Hospital cohort exhibiting inconclusive and/or equivocal Sanger Sequencing results were also tested with the mutation-specific restriction enzyme digestion assay as reported previously.13 PCR products were used to screen for the two previously reported GNAS codons p.R201 and p.Q227 mutations in which 10 substitutions are reported to occur. In brief, for five of the mutations (p.R201G, p.R201S, p.Q227R, p.Q227H, and p.Q227K) the nucleotide substitution directly altered the cutting site of the restriction enzyme. For each of the other five mutations (p.R201C, p.R201H, p.R201P, p.R201L, and p.Q227L), a mismatch primer was designed containing a specific nucleotide substitution allowing size discrimination between alleles containing the wild type or mutant nucleotide following digestion of PCR products with the appropriate restriction enzyme.13 Products were separated through 8% polyacrylamide gels and visualized using the Bio-Rad Gel Doc 2000TM system (Bio-Rad Laboratories, Hercules, CA, USA).
Assessment of Amplifiable DNA by Droplet Digital PCR
To assess the number of amplifiable DNA molecules of the 36 parosteal osteosarcoma samples from the Royal National Orthopaedic Hospital cohort, we performed droplet digital PCR on the Q × 200 droplet digital PCR system (Bio-Rad Laboratories). The reaction was performed in duplicate with 25 ng of DNA and tested using 0.1 μM of the GNAS p.R201 exon 8 primer (Supplementary Table 1) and EvaGreen Supermix (Bio-Rad Laboratories) in order to count the number of amplifiable DNA molecules that would have been added to the Fluidigm next generation sequencing library preparation. Highly purified Human Genomic DNA (Bioline Reagents, London, UK) was used as a positive control.
Results
MDM2 Amplification by Fluorescent In Situ Hybridization
Table 2 shows the percentage of MDM2 amplifications in low-grade osteosarcomas. Of the informative samples analyzed by fluorescent in situ hybridization from the Royal National Orthopaedic Hospital tumor set, MDM2 amplification was detected in 28/34 (82%) parosteal osteosarcomas, in 2/5 (40%) low-grade central osteosarcomas, and in none of the 6 periosteal osteosarcomas. Of the 24 informative samples from the University Hospital Basel, 6/9 (67%) parosteal osteosarcomas, 3/12 (25%) low-grade central osteosarcomas, and none of three periosteal osteosarcomas showed MDM2 amplification. Fluorescent in situ hybridization analysis of the seven parosteal osteosarcoma cases, from the Royal National Orthopaedic Hospital, with conventional and dedifferentiated components revealed MDM2 amplification in both components.
Next Generation Sequencing
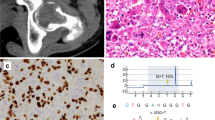
GNAS substitutions involving p.R201, p.Q227, and p.K233 codons were not found in 47 samples from 40 patients with osteosarcoma from the Royal National Orthopaedic Hospital cohort that were analyzed by next generation sequencing (Table 3). Seven of these samples represented the dedifferentiated component of a matching conventional parosteal osteosarcoma (Figures 1b and c). All cases, apart from one, generated reads at high depth, averaging 2800 filtered reads (Figures 1b and c), with the outlying case generating only 523 reads. All six GNAS positive controls (p.R201C and p.R201H) generated an average of 3200 filtered reads and revealed the relevant mutations (Figure 1a).

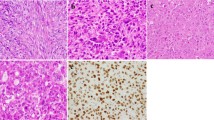
Mutational analyses of GNAS in parosteal osteosarcoma and fibrous dysplasia by next generation sequencing and Sanger sequencing. (a) Haematoxylin and eosin (H&E) image of a fibrous dysplasia (original magnification: × 20). (a1) Representation of the reads aligned to the reference genome (Integrative Genomics Viewer software—IGV v 2.1, Broad Institute) for a GNAS mutation p.R201C (CGT>TGT) in exon 8 in a fibrous dysplasia. (a2) Sanger sequencing chromatogram of a GNAS mutation p.R201C (CGT>TGT) in a fibrous dysplasia. (b) H&E image of a conventional parosteal osteosarcoma (original magnification: × 20). (b1) Representation of the reads aligned to the reference genome for a GNAS exon 8 wild-type in a conventional parosteal osteosarcoma. (b2) Sanger sequencing chromatogram of a GNAS exon 8 wild-type in a conventional parosteal osteosarcoma. (c) H&E image of a dedifferentiated parosteal osteosarcoma (original magnification: × 20). (c1) Representation of the reads aligned to the reference genome for a GNAS exon 8 wild-type in a dedifferentiated parosteal osteosarcoma. (c2) Sanger sequencing chromatogram of a GNAS exon 8 wild-type in a dedifferentiated parosteal osteosarcoma.
Sanger Sequencing
Bidirectional Sanger sequencing was performed for GNAS codons p.R201 and p.Q227: mutations were not detected in any of the 57/62 parosteal (p.R201) and in 58/62 parosteal (p.Q227) osteosarcoma samples from 55 individuals as well as in none of the included 24 low-grade central osteosarcomas and 11 periosteal osteosarcomas (combined from both centers). In total, 20 cases exhibited inconclusive sequencing results by Sanger sequencing, 14 cases from the University Hospital Basel and 6 cases from the Royal National Orthopaedic Hospital. The six samples from the Royal National Orthopaedic Hospital were re-sequenced in both directions by Sanger sequencing, after which five of six samples provided interpretable calls in both directions revealing the absence of GNAS mutations. These results were further validated by demonstrating failure to detect a GNAS substitution in these 5/6 samples by mutation-specific restriction enzyme digestion (data not shown). The only Royal National Orthopaedic Hospital case that failed to give a conclusive result by Sanger sequencing and/or mutation-specific restriction enzyme digestion was a parosteal osteosarcoma for which the DNA revealed low depth of coverage by next generation sequencing (523 reads) and it was also noteworthy that this case revealed poor quality DNA (vide infra—Assessment of Amplifiable DNA by Droplet Digital PCR) and was therefore excluded from the analysis. Sanger sequencing of codon p.R201 and codon p.Q227 failed in eight and six University Hospital Basel cases, respectively (Table 4). These were not analyzed by mutation-specific restriction enzyme digestion.
Assessment of Amplifiable DNA by Droplet Digital PCR
Assessment of the quality of amplifiable DNA of the parosteal osteosarcomas from the Royal National Orthopaedic Hospital was undertaken by droplet digital PCR. We considered any sample with at least 100 amplifiable copies of the GNAS exon 8 region per next generation sequencing run to be satisfactory. The only case to have <100 amplifiable copies was the parosteal sample that failed both bidirectional Sanger sequencing and mutation-specific restriction enzyme digestion (data not shown).
Discussion
In this study, we assembled a large series of low-grade osteosarcomas from two independent institutions and failed to detect a single GNAS mutation in 55 parosteal, 24 low-grade central, and 11 periosteal osteosarcomas. All cases were thoroughly re-evaluated at a histological level by experienced bone tumor pathologists and the diagnosis was confirmed. In addition, fluorescent in situ hybridization analyses revealed 34/43 (79%) informative parosteal osteosarcomas to harbor MDM2 amplifications which is in line with previous reports.5 Bidirectional Sanger sequencing was used as the ‘gold standard’ for GNAS mutational analysis as this technology has been ‘tried and tested’ for many years. Furthermore, Carter et al.22 had employed this technique to detect GNAS mutations in 5/9 parosteal osteosarcomas.22 The lack of GNAS mutations detected by Sanger sequencing was confirmed in 46 osteosarcoma samples analyzed by next generation sequencing—a more sensitive technology—whereas in the positive controls the mutations were identified. Furthermore, the absence of specific mutations was confirmed by mutation-specific restriction enzyme digestion as a third technology in a smaller subset of cases.
Of the five cases of parosteal osteosarcomas that Carter and colleagues found to harbor a GNAS mutation, three dedifferentiated during the course of the disease. Interestingly, the mutations were detected in both the conventional and dedifferentiated components in one case, only in the conventional component in another case, and only in the dedifferentiated component in a third case.22 We therefore analyzed both the conventional and dedifferentiated components of seven dedifferentiated parosteal osteosarcomas for the presence of GNAS mutations but failed to detect any GNAS mutation. The periosteal osteosarcomas included in our study neither showed GNAS mutations nor MDM2 amplifications. Although periosteal and parosteal osteosarcomas both develop on the surface of bones and belong to the group of juxtacortical osteosarcomas, periosteal osteosarcomas do not share MDM2 amplifications as a common feature which is line with previous reports.25
DNA isolated from formalin-fixed paraffin-embedded tissue is often highly fragmented and chemically degraded, especially in decalcified samples. As this might also explain our failure to detect GNAS mutations, we tested a subset of our samples for DNA quality: the DNA was found to be of sufficient quality in all but a single case that was excluded from the analysis. Consequently, the absence of GNAS mutations in low-grade osteosarcomas cannot be attributed to the lack of amplifiable DNA.
The absence of GNAS alterations in 24 low-grade central osteosarcomas (5/17 with MDM2 amplification) provides strong evidence that these mutations are mutually exclusive in fibrous dysplasia and low-grade central osteosarcoma. However, GNAS mutations per se do not exclude malignancy as malignant transformation of fibrous dysplasia can rarely occur, especially in polyostotic disease. However, in the few reported cases and in our experience (not reported), malignant transformation of fibrous dysplasia usually shows high-grade morphology and does not raise diagnostic difficulties. Hence, interpretation of genetic alterations must always be performed in the context of morphology and imaging.26, 27
Our findings support prior observations that in the appropriate context, the detection of a GNAS mutation in a primary bone tumor is supportive of a diagnosis of fibrous dysplasia, and GNAS mutations rarely, if ever, occur in parosteal osteosarcoma. The reason for the findings by Carter and colleagues, however, remain elusive. DNA contamination in a laboratory is always a concern when using PCR-based technologies and strict controls and practices must be adhered to. An alternative explanation is that the DNA was degraded and a PCR artifact occurred. On the basis of this study, we argue that the detection of GNAS mutations still remains a useful molecular tool for distinguishing fibrous dysplasia from low-grade osteosarcoma.
References
Hang JF, Chen PC . Parosteal osteosarcoma. Arch Pathol Lab Med 2014;138:694–699.
Dorfman HD, Ishida T, Tsuneyoshi M . Exophytic variant of fibrous dysplasia (fibrous dysplasia protuberans). Hum Pathol 1994;25:1234–1237.
Yoshida A, Ushiku T, Motoi T et al. Immunohistochemical analysis of MDM2 and CDK4 distinguishes low-grade osteosarcoma from benign mimics. Mod Pathol 2010;23:1279–1288.
Wunder JS, Eppert K, Burrow SR et al. Co-amplification and overexpression of CDK4, SAS and MDM2 occurs frequently in human parosteal osteosarcomas. Oncogene 1999;18:783–788.
Dujardin F, Binh MB, Bouvier C et al. MDM2 and CDK4 immunohistochemistry is a valuable tool in the differential diagnosis of low-grade osteosarcomas. Mod Pathol 2011;24:624–637.
Duhamel LA, Ye H, Halai D et al. Frequency of Mouse Double Minute 2 MDM2 and Mouse Double Minute 4 MDM4 amplification in parosteal and conventional osteosarcoma subtypes. Histopathology 2012;60:357–359.
Bertoni F, Bacchini P, Staals EL et al. Dedifferentiated parosteal osteosarcoma: the experience of the Rizzoli Institute. Cancer 2005;103:2373–2382.
Sheth DS, Yasko AW, Raymond AK et al. Conventional and dedifferentiated parosteal osteosarcoma: diagnosis, treatment, and outcome. Cancer 1996;78:2136–2145.
Okada K, Frassica FJ, Sim FH et al. Parosteal osteosarcoma: a clinicopathological study. J Bone Joint Surg Am 1994;76:366–378.
Tabareau-Delalande F, Collin C, Gomez-Brouchet et al. Diagnostic value of investigating GNAS mutations in fibro-osseous lesions a retrospective study of 91 cases of fibrous dysplasia and 40 other fibro-osseous lesions. Mod Pathol 2013;26:911–921.
Kashima TG, Gamage NM, Ye H et al. Locally aggressive fibrous dysplasia. Virchows Arch 2013;463:79–84.
Liang Q, Wei M, Hodge L et al. Quantitative analysis of activating alpha subunit of the G protein Gsα mutation by pyrosequencing in fibrous dysplasia and other bone lesions. J Mol Diagn 2011;13:137–142.
Idowu BD, Al-Adnani M, O’Donnell P et al. A sensitive mutation-specific screening technique for GNAS1 mutations in cases of fibrous dysplasia the first report of a codon 227 mutation in bone. Histopathology 2007;50:691–704.
Pollandt K, Engels C, Kaiser E et al. Gsalpha gene mutations in monostotic fibrous dysplasia of bone and fibrous dysplasia-like low-grade central osteosarcoma. Virchows Arch 2001;439:170–175.
Toyosawa S, Yuki M, Kishino M et al. Ossifying fibroma vs fibrous dysplasia of the jaw molecular and immunological characterization. Mod Pathol 2007;20:389–396.
Shi RR, Li XF, Zhang R et al. GNAS mutational analysis in differentiating fibrous dysplasia and ossifying fibroma of the jaw. Mod Pathol 2013;8:1023–1031.
Candeliere GA, Roughley PJ, Glorieux FH . Polymerase chain reaction-based technique for the selective enrichment and analysis of mosaic arg201 mutations in Gas, from patients with fibrous dysplasia of bone. Bone 1997;21:201–206.
Riminucci M, Fisher LW, Majolagbe et al. Clinical vignette: a novel GNAS1, R201G, in McCune–Albright syndrome. J Bone Miner Res 1999;14:1987–1989.
Landis CA, Masters SB, Spada et al. GTPase inhibiting mutations activate the a chain of Gs and stimulate adenylate cyclase in human pituitary tumours. Nature 1989;340:692–696.
Gorelov VN, Dumon K, Barteneva NS et al. Overexpression of Gs alpha subunit in thyroid tumors bearing a mutated Gs alpha gene. J Cancer Res Clin Oncol 1995;121:219–224.
Nishikawa G, Sekine S, Ogawa R et al. Frequent GNAS mutations in low-grade appendiceal mucinous neoplasms. Br J Cancer 2013;4:951–958.
Carter JM, Inwards CY, Jin L et al. Activating GNAS mutations in parosteal osteosarcoma. Am J Surg Pathol 2014;38:402–409.
Going JJ, Lamb RF . Practical histological microdissection for PCR analysis. J Pathol 1996;179:121–124.
Kashima T, Halai D, Ye H et al. Sensitivity of MDM2 amplification and unexpected multiple faint alphoid 12 (alpha 12 satellite sequences) signals in atypical lipomatous tumor. Mod Pathol 2012;10:1384–1396.
Righi A, Gambarotti M, Benini S et al. MDM2 and CDK4 expression in periosteal osteosarcoma. Hum Pathol 2015;46:549–553.
Mardekian SK, Tuluc M . Malignant sarcomatous transformation of fibrous dysplasia. Head Neck Pathol 2015;9:100–103.
Hatano H, Morita T, Ariizumi T et al. Malignant transformation of fibrous dysplasia: a case report. Oncol Lett 2014;8:384–386.
Acknowledgements
Funding was provided by Skeletal Cancer Action Trust. Support was provided to Adrienne Flanagan by the NIHR, the University College London Hospitals Biomedical Research Centre, and the Cancer Research UK University College London Experimental Cancer Medicine Centre. The Stanmore biobank is supported by the Research and Development Department of the Royal National Orthopaedic Hospital. Carolina Salinas-Souza was funded by São Paulo Research Foundation FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo) project number 2013/13573-9. Daniel Baumhoer and Michal Kovac were supported by the foundation for the preservation of the Basel Bone Tumor Reference Centre.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Supplementary information
Rights and permissions
About this article

Cite this article
Salinas-Souza, C., De Andrea, C., Bihl, M. et al. GNAS mutations are not detected in parosteal and low-grade central osteosarcomas. Mod Pathol 28, 1336–1342 (2015). https://doi.org/10.1038/modpathol.2015.91
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2015.91
This article is cited by
-
What’s new in bone forming tumours of the skeleton?
Virchows Archiv (2020)
-
Osteoidbildende Knochentumoren
Der Pathologe (2020)
-
Molekulare Pathologie in der Diagnostik der Knochentumoren: Aktueller Stand
Der Pathologe (2020)
-
The curious case of Gαs gain-of-function in neoplasia
BMC Cancer (2018)
-
Fibrous dysplasia: an unusual case of a very aggressive form with costo-vertebral joint destruction and invasion of the contralateral D7 vertebral body
Skeletal Radiology (2018)
