
Abstract
Embryonal rhabdomyosarcoma of the uterine cervix is an uncommon presentation of the most common soft-tissue sarcoma in the first decades of life. Unlike embryonal rhabdomyosarcoma in other anatomic sites, in which 70–80% of cases present before 9 years of age, the average age in our series of 14 cervical cases was 12.4 years (median, 13 years), with an age range of 9 months to 32 years at diagnosis. Of the 14 cases, 12 presented as a polyp at the cervical os; two patients had an infiltrative mass in the cervix without a botryoid polyp. The polyps measured 1.5–5 cm and all had the histopathological pattern of the sarcoma botryoides variant of embryonal rhabdomyosarcoma, with condensations of primitive and differentiated rhabdomyoblasts beneath the surface epithelium and around endocervical glands. Nodules of benign-appearing cartilage were present in the stroma of six cases (43%). One of the embyronal rhabdomyosarcomas from the youngest patient, 9 months old, also had a distinctive microscopic focus of immature tubular profiles in a primitive stroma; these tubules expressed epithelial and neuroendocrine markers. Two patients had a pleuropulmonary blastoma, one diagnosed 9 years before the embryonal rhabdomyosarcoma of the cervix and the other recognized synchronously. This latter 9-year old had a DICER1 germline mutation. One patient presented with hirsutism and had a Sertoli–Leydig cell tumor, an incidentally detected cervical embryonal rhabdomyosarcoma, and nodular hyperplasia of the thyroid. Although a pleuropulmonary blastoma was not documented in the latter patient, ovarian sex-cord stromal tumors and nodular hyperplasia of the thyroid are manifestations of the pleuropulmonary blastoma family tumor and dysplasia syndrome (OMIM 601200). Embryonal rhabdomyosarcoma of the cervix must be distinguished from other rare entities, including adenosarcoma, malignant mixed Mullerian tumor and low-grade stromal sarcoma, as the former has a better prognosis; 12 of our 14 patients remain disease-free following conservative surgery and chemotherapy. Our study suggests that cervical embryonal rhabdomyosarcoma may be another pathological manifestation in the spectrum of extrapulmonary pathology in the setting of pleuropulmonary blastoma.
Similar content being viewed by others
Main
Rhabdomyosarcoma is the most common soft-tissue sarcoma of childhood, accounting for 50% of those cases diagnosed at or before 20 years of age.1 Unlike soft-tissue sarcomas in adults, with a predilection for the extremities and retroperitoneum, rhabdomyosarcomas in children preferentially occur in the head and neck region (35%) and genitourinary tract (25%), with the bladder, prostate and vagina as the most common sites.2 Rhabdomyosarcomas arising in the genitourinary tract are commonly the sarcoma botryoides variant and are typically seen in the first decade of life—often before 3 years of age in the case of vaginal rhabdomyosarcoma.3, 4 In the adolescent and young adult population (up to 30 years of age), rhabdomyosarcoma accounts for approximately 30% of soft-tissue sarcomas, but in older patients both embryonal rhabdomyosarcoma and alveolar rhabdomyosarcomas are very uncommon to rare.5, 6
One of the least common sites for rhabdomyosarcoma in the genitourinary tract is the uterine cervix, but its occurrence has been well documented in the study of 13 cases by Daya and Scully.7 The subsequent literature on cervical rhabdomyosarcoma has been entangled with another neoplasm of the cervix, the adenosarcoma; the report by Bagga and co-workers8 of a cervical adenosarcoma in a 15-year-old female patient is illustrative of the confusion between these two neoplasms.
This report of 14 cases of cervical rhabdomyosarcoma in individuals between the ages of 9 months and 32 years at the time of diagnosis demonstrates some of the unique clinical and pathological aspects of this neoplasm, including its association with extrauterine pathology, possibly linking it with the familial pleuropulmonary blastoma tumor predisposition and dysplasia syndrome.9
Materials and methods
The 14 cases in this study were retrieved from the files of the Lauren V Ackerman Laboratory of Surgical Pathology, Barnes-Jewish and St Louis Children's Hospitals, Washington University Medical Center (St Louis, MO, USA), utilizing the search terms ‘uterus and rhabdomyosarcoma’ and ‘uterine cervix and rhabdomyosarcoma’. A total of 26 cases were identified with these search terms during the period from 1990 to 2010 and 14 cases of rhabdomyosarcoma confined to the cervix were found in individuals between the ages of 9 months and 32 years at diagnosis. The remaining 12 cases were women between the ages of 47 and 87 years who had pure rhabdomyosarcomas of the uterine fundus and cervix (four cases), malignant mixed Mullerian tumors with heterologous rhabdomyosarcoma (seven cases) and adenosarcoma with rhabdomyosarcoma overgrowth (one case) of the uterine fundus. Eight cases in this study were diagnosed and treated in this medical center and the remaining cases were submitted in consultation from other institutions. One of the latter cases had been referred to the International Pleuropulmonary Blastoma Registry through one of the authors (DAH). Follow-up was obtained from the referring pathologists in the consultation cases (available in five of six cases) and from the treating surgeon and/or oncologist in those cases from this medical center.
All available microscopic sections and available medical records were reviewed. Immunohistochemistry was performed on 14 cases; all were stained for vimentin (V9, mouse) and desmin (DE-9-11, mouse), and five for myogenin (FSD, mouse). The one Sertoli–Leydig cell tumor was stained for vimentin, inhibin (RI, mouse) and WT-1 (6F-H2, mouse). All antibodies (predilute) were obtained through Ventana Medical System (Tucson, AZ, USA).
This study was approved by the Institutional Review Board at Washington University in St Louis under the provisions of an ongoing Pleuropulmonary Blastoma Study.
Results
Clinical History, Findings and Follow-Up
The 14 patients ranged in age from 9 months to 32 years at the time of diagnosis, with a mean age of 12.4 years and a median age of 13 years (Table 1). With the exception of four cases (ages 9 months, 3 years, 5 years and 7 years), the remaining 10 patients were 13 years old or older at diagnosis.
Vaginal bleeding in the form of ‘spotting’ in the four premenarchal children had durations of a week or less in contrast to a history of extramenstrual spotting or bleeding for ‘several’ weeks in the postmenarchal female patients. One patient (Case 14), a 32-year-old female, was thought initially to have a prolapsed myoma when she presented with heavy vaginal bleeding, tachycardia and anemia. An asymptomatic cervical polyp was detected incidentally in a 13-year-old patient (Case 4) at the time of her clinical presentation with hirsutism; this same patient also had a right adnexal mass and an enlarged, multinodular goiter. The additional clinical history in a 12-year-old patient (Case 12) was the resection of the right lower lobe at the age of 3½ years for a ‘rhabdomyosarcoma’ of the lung; this neoplasm was examined in the course of this study and found to be a solid or type III pleuropulmonary blastoma. There was a disease-free interval of almost 8 years before the detection of the cervical polyp in this patient.
A ‘vaginal’ polyp was detected in each case, except for two patients, one a 15-year-old girl (Case 8) with a firm mass of the cervix as well as urinary tract obstruction because of pelvic mass effect. The other patient, a 32-year-old woman (Case 14), had a hemorrhagic and necrotic polypoid mass arising from the endocervical canal without accompanying fundic enlargement; a prolapsed leiomyoma was the initial clinical impression. The polyps in the other 12 patients were described as arising from the region of the cervical os, measuring 1.5–5 cm and having a pedunculated appearance and smooth glistening mucosal surfaces with focal hemorrhage.
In total, 12 patients had polypectomies without any preoperative suspicion of malignancy. The two remaining patients (Cases 8 and 14) had only biopsies initially. Following the diagnosis of rhabdomyosarcoma in all but Case 8, additional surgical procedures included cervical biopsies to detect any residual tumor, and loop electrosurgical excision procedures with or without complete conization. Adjuvant chemotherapy consisting of vincristine, actinomycin and cyclophosphamide (VAC) was administered to 13 patients (Table 1). Upon completion of the VAC therapy, repeat biopsies were performed to ascertain the presence of residual tumor. A local recurrence developed in a 13-year-old female patient (Case 5) after an 8-week course of chemotherapy. Two patients, 5 and 15 years old (Cases 2 and 8), had radical hysterectomies after local recurrence or persistence of tumor after chemotherapy. One patient (Case 14) elected to have only follow-up biopsies and curettages following the resection of an 8.7 cm polypoid mass from the cervix; she has remained tumor-free for 6 years after the diagnosis without additional treatment including chemotherapy, though counseled otherwise. The 13-year-old female patient (Case 4) with the Sertoli–Leydig cell tumor of the right ovary additionally had a right salpingo-oophorectomy and an abdominal staging procedure that revealed no evidence of peritoneal spread of either the ovarian or cervical neoplasm; she also had a total thyroidectomy that revealed multinodular hyperplasia. During the course of pre-therapy staging in one patient (Case 11), a large cyst with delicate septations was discovered in the right upper lobe of the lung and was resected.
No tumor-related deaths have been recorded to date, but two patients have active disease, one with recurrent rhabdomyosarcoma in the pelvis after a radical hysterectomy (Case 8) and the other with recurrent Sertoli–Leydig cell tumor approximately 7 years after the original diagnosis (Case 4). One patient (Case 5) has been lost to follow-up, but was known to have recurrent rhabdomyosarcoma in the cervix 1 year after completing the initial course of chemotherapy. Eight patients with 2 years or more of follow-up have remained tumor-free from 2 to 20 years after the initial diagnosis of rhabdomyosarcoma (Table 1). The most common therapeutic approach was local excision of the tumor, VAC chemotherapy and biopsies of the cervix upon completion of chemotherapy for evidence of residual disease.
Pathological and Immunohistochemical Findings
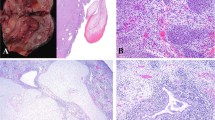
Of the 14 cervical tumors, 12 had similar gross findings of a polyp(s) measuring 1.5–5 cm in greatest dimension and attached to the cervix in or near the os by a narrow pedicle. The external surfaces were described as glistening, with apparent superficial mucosal ulcerations or erosions in some cases. One tumor was a 6–7 cm mass with ulceration and necrosis that arose from the cervix and partially filled the vagina (Case 8). Another tumor (Case 14) was an 8.7 × 6.2 × 5.3 cm3 (144 g), gelatinous, pale pinkish-tan, myxoid and focally hemorrhagic polypoid mass rather than a smooth-surfaced pedunculated polyp.
Microscopically, the polypoid tumors were characterized by a continuous or discontinuous band of small primitive or immature rhabdomyoblastic cells beneath an intact squamous mucosa, but the tumor cells were less apparent when the surface epithelium was eroded and hemorrhage was present in the underlying stroma (Figure 1a). The cambium layer of subepithelial tumor cells was more easily identified in the less inflamed and ulcerated areas of the polyp (Figure 1b). A pale-staining, hypocellular stroma with a myxoid appearance characterized the central portion of the polyps. Glandular structures lined by a mucinous and/or non-mucinous columnar epithelium were identified in all cases; these benign-appearing glands were present in varying numbers from one microscopic field to another and were randomly distributed throughout the polyp (Figure 2a and b). Another feature was the presence of one or more nodules of cartilage in six of 14 cases (43%); these nodules varied in cellularity and nuclear atypia, but none had overtly sarcomatous features (Figure 3a and b). Other than the cambium layer of rhabdomyoblasts beneath the mucosal surface, condensations of similar appearing primitive cells were present uniformly around the glands (Figure 4). None of the tumors had a spindle cell sarcomatous pattern resembling fibrosarcoma or endometrial stromal sarcoma.

(a) Embryonal rhabdomyosarcoma of the cervix in a 13-year-old female patient (Case 9) shows erosion of the surface epithelium and interstitial hemorrhage, which can obscure the underlying pathology. (b) Contiguous areas of the same polyp with intact surface epithelium demonstrated to better advantage the primitive rhabdomyoblasts.

(a) This 9-year-old female (Case 11) presented with a ‘vaginal polyp’, which was traced into the endocervix. Adjacent endocervical glands are surrounded in part by subepithelial tumor cells. Note that the stroma beyond the glands has a hypocellular myxoid appearance. (b) The rhabdomyoblastic nature of the tumor cells is noted by the small-rounded to spindle-shaped tumor cells with delicate tails of eosinophilic cytoplasm.

The nodules of cartilage varied in size and degree of cellularity. (a) This nodule of cartilage was one of several within the stroma (Case 11). The adjacent endocervical gland provides some perspective on its size. (b) A smaller, more cellular nodule is adjacent to an endocervical gland with accompanying embryonal rhabdomyosarcoma.

The dense circumferential pattern of rhabdomyoblasts around endocervical glands was a consistent feature in these polypoid neoplasms (Case 9), regardless of age at diagnosis.
Three of the cervical neoplasms displayed some histological features that were unique to them in contrast to the other 11 cases. One of these tumors occurred in the youngest patient in our series, who was 9 months old at diagnosis (Case 9). This tumor was a polyp with the features of a botryoid embryonal rhabdomyosarcoma, but in addition had a well-circumscribed island of primitive tubular structures in a background of immature non-rhabdomyoblastic mesenchyme (Figure 5a and b). The second case, a 15-year-old girl (Case 8), presented with a mass rather than as a polyp. This neoplasm had a variety of histological patterns, including the condensation of primitive rhabdomyoblasts around endocervical glands, foci of spindled rhabdomyoblasts, islands of crowded primitive small cells with a blastemal appearance and scattered aggregates of larger anaplastic-appearing tumor cells (Figure 6a–d). The third neoplasm in the oldest of the 14 patients, a 32-year-old female (Case 14), was a hemorrhagic, polypoid mass with the periglandular pattern of rhabdomyoblasts and areas of deep tumor invasion into the smooth muscle of the cervix (Figure 7a and b). This patient was managed by a partial resection of the cervix without adjuvant chemotherapy. She has had follow-up cervical biopsies and cytologies following the resection and has remained tumor-free for 6 years.

(a) A distinct focus of tumor was present in a sarcoma botryoides of the cervix in a 9-month-old female patient (Case 9). The embryonal rhabdomyosarcomatous pattern is depicted in part in Figure 4. (b) The primitive tubular profiles are surrounded by smaller rosette-like structures in a background of immature, non-rhabdomyosarcomatous mesenchyme.

One case presented as a cervical mass rather than a polyp in a 15-year-old female patient (Case 8). This neoplasm displayed a variety of patterns. (a) An endocervical gland is surrounded in part by embryonal rhabdomyoblasts. (b) Other foci were represented by spindle cell rhabdomyosarcoma. (c) Yet another pattern of this neoplasm had a more blastematous appearance. (d) Anaplastic rhabdomyoblasts were found in several foci. The composite patterns in this neoplasm have a similarity to the histological collage, which is seen in the solid areas of the pleuropulmonary blastoma.

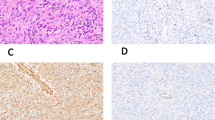
Embryonal rhabdomyosarcoma presented as a hemorrhagic, necrotic polyp from the cervix of this 32-year-old female patient (Case 14). (a) This tumor displayed a range of features from primitive rhabdomyoblasts to well-differentiated rhabdomyoblasts with myotube formation. (b) Pleomorphic tumor cells accompanied rhabdomyoblasts in areas of infiltration into the smooth muscle of cervix. (c) Desmin expression was consistently expressed in the primitive rhabdomyoblasts and differentiated myotubes, but less consistently in the large pleomorphic tumor cells. (d) Nuclear positivity for myogenin was seen in rhabdomyoblasts, but infrequently in the pleomorphic cells.
Three of the cases had additional extrauterine pathology, pleuropulmonary blastoma in two cases (Cases 11 and 12) and Sertoli–Leydig cell tumor of the right ovary as well as nodular hyperplasia of the thyroid in the third patient (Case 4). A solid or type III pleuropulmonary blastoma was diagnosed in the right lung at 3.5 years of age in one patient (Case 12); this tumor displayed the complex sarcomatous pattern of embryonal rhabdomyosarcoma, high-grade spindle cell sarcoma, blastema-like nests and anaplasia. The other pleuropulmonary blastoma was detected in the right upper lobe, during the imaging phase of staging after the diagnosis of embryonal rhabdomyosarcoma (Case 11; Figure 8a). A right upper lobe resection yielded a multicystic lesion measuring 7.5 × 6.5 × 6 cm3 that replaced almost the entire lobe (Figure 8b). The septa of this multiloculated cyst were variably thickened and had a generally hypocellular stroma. Extensive sampling failed to identify primitive small cells or rhabdomyoblasts beneath the cuboidal epithelial lining. Nodules of cartilage were not identified. The features of the multiloculated cyst were those of an involuted or ‘regressed’ pleuropulmonary blastoma (Figure 9a and b). Additional studies were performed in this child and she was found to have a germline DICER1 mutation, c5104CST, which she had inherited from her mother. In the third case (Case 4), the right ovary weighed 97 g and had a yellowish-tan, mucoid cut surface with focal areas of hemorrhage. Densely cellular foci composed of grouped tubular profiles and small strands of Leydig cells were separated by a loose mesenchyme as features of a Sertoli–Leydig cell tumor (Figure 10a and b).

(a) A large multicystic lesion in the right upper lobe was discovered during the non-invasive staging in this 9-year-old female patient (Case 11). (b) The resected lobe contained a 7.5 × 6.5 × 6 cm3 multicystic lesion, which is seen in the unopened specimen before its collapse after the release of clear fluid. The septa had a smooth translucent appearance and no areas of gross thickening or masses were identified.

(a) Cystic pleuropulmonary blastoma (type I) was composed of cysts, some with a delicate thread-like quality and others with a more prominent fibrous stroma. (b) The cysts were lined by a low cuboidal epithelium and a pale hypocellular stroma. Condensations of primitive round cells or rhabdomyoblasts were not identified after extensive sampling.

(a) Sertoli–Leydig cell tumor of the right ovary shows the presence of discrete solid nests in a hemorrhagic background. (b) Elsewhere, cords of Leydig cells are present in the fibromyxoid stroma.
Immunohistochemistry for vimentin and desmin was performed on each of the cervical tumors. The primitive small cells in the cambium layer and around the glands were immunoreactive for desmin, whereas the stroma throughout displayed vimentin positivity (Figure 11a and b). The one non-botryoid rhabdomyosarcoma of the cervix demonstrated desmin reactivity throughout this solid multipatterned neoplasm (Case 8). Nuclear reactivity for myogenin was demonstrated in five cases with available material (Cases 8–11). The distinctive focus composed of immature mesenchyme with the primitive tubules in the cervical polyp from the 9-month-old patient (Case 9), which revealed that the tubular structures were positive for pancytokeratin, chromogranin and neuron-specific enolase, whereas the background stroma was reactive for vimentin, but negative for desmin and myogenin (Figure 12a and b). The portion of the polyp with embryonal rhabdomyosarcoma revealed desmin and myogenin (nuclear reactivity) positivity in the population of primitive rhabdomyoblasts around the endocervical glands (Figure 12c and d). The Sertoli–Leydig cell tumor was immunoreactive for vimentin, inhibin and WT-1 (Case 4).

This tumor occurrence in the oldest patient in this series, a 32-year-old woman (Case 14). (a) A mantle of desmin-positive rhabdomyoblasts was surrounding an endocervical gland. (b) Nuclear staining for myogenin was present in most, but not all, rhabdomyoblasts, which is relatively common in embryonal rhabdomyosarcoma.

This polypoid lesion presented in the cervix in a 9-month-old female (Case 9), the youngest patient in this series. (a) The discrete island of tubules and mesenchyme in this neoplasm contains tubular profiles, which are pancytokeratin positive. (b) Many of these same tubular structures are neuron-specific enolase (shown) and synaptophysin positive. (c) All of the primitive small tumor cells are positive for vimentin and a fraction are positive for desmin (shown). (d) Nuclear positivity for myogenin is seen in a population of primitive small cells without obvious rhabdomyoblastic differentiation.
Discussion
Embryonal rhabdomyosarcomas of the uterine cervix are rare tumors with unique clinical and pathological findings and associations that distinguish them from embryonal rhabdomyosarcomas occurring in other sites. These tumors have been mainly discussed in the context of individual case studies, except for a single earlier study of 13 cases.7 Almost 70% of all embryonal rhabdomyosarcomas are diagnosed before 20 years of age, with 40% of cases occurring between 0 and 4 years of age; the average age at diagnosis of embryonal rhabdomyosarcoma of the vagina is approximately 2 years old.1, 3, 4, 10 However, cervical embryonal rhabdomyosarcomas typically occur in older patients. Our series reflected a mean and median age at diagnosis of 13 years, similar to the study of Daya and Scully (mean=18 years; median=16 years).7
Like other examples of embryonal rhabdomyosarcoma arising in organs and anatomic sites with access to a space as in the nasopharynx, middle ear, common bile duct, bladder and vagina, in most cases the cervical embryonal rhabdomyosarcoma has the gross and microscopic features of the sarcoma botryoides variant.11, 12, 13 This was true in 12 of 14 cases in this study, where these neoplasms grew into the vaginal vault from the cervix and had microscopically evident cambium layers of malignant cells underlying the surface epithelium. The other two cases (Cases 8 and 14) presented as infiltrative masses rather than polyps and had complex patterns with focal areas of embryonal rhabdomyosarcoma with a periglandular orientation of small primitive rhabdomyoblasts, and additionally other patterns of spindle cell rhabdomyosarcoma and anaplasia.
Nodules of cartilage were identified in the stroma in six cases, including Case 8 with spindle cell and anaplastic features. The cartilage was a minor component in each of these six cases and was cellular and immature, but not with overtly malignant features, distinct in appearance from the chondrosarcomatous areas of a malignant mixed Mullerian tumor with heterologous sarcomatous elements. The presence of cartilaginous nodules was first noted in six (45%) of 13 embryonal rhabdomyosarcomas of the cervix by Daya and Scully.7 There is also a report of a primary embryonal rhabdomyosarcoma of the ovary, with a focus of ‘fetal-type cartilage’ in a 25-year-old woman;14 as judged by the photomicrographs of the latter tumor, it is very similar to the six cervical embryonal rhabdomyosarcomas with heterologous cartilage in this study.
One of our 14 cases with the sarcoma botryoides pattern, occurring in the 9-month-old infant, contained a well-defined island of tumor, distinct from the embryonal rhabdomyosarcoma, which was composed of immature tubules resembling primitive neural tubes in a background of primitive, non-rhabdomyosarcomatous mesenchyme. These tubular structures were immunoreactive for cytokeratin, neuron-specific enolase and chromogranin. There was no evidence of small strands and cords of tumor cells in a myxoid background that would suggest a yolk sac carcinoma. We chose to interpret this focus as ‘teratoid’ without a ready explanation for its presence. The coincidence of rhabdomyosarcoma and a primitive neuroectodermal or neuroepithelial component is found rarely in germ cell neoplasms and in the so-called malignant ectomesenchymoma.15, 16, 17 Rhabdomyosarcomatous elements are also present in the malignant triton tumor. Neither the nodules of cartilage nor the primitive neuroectodermal focus were thought to fulfill the morphological criterion for a malignant mixed Mullerian tumor with both malignant epithelial and malignant mesenchymal components.
As noted earlier, the literature on the topic of embryonal rhabdomyosarcoma and adenosarcoma of the cervix highlights the historical difficulties in differentiating between these two neoplasms. An essential distinguishing characteristic is the exclusive presence of embryonal rhabdomyosarcoma in the cervical embryonal rhabdomyosarcoma and, in most cases, the gross and microscopic features of sarcoma botryoides.18 In the case of adenosarcoma, the sarcomatous component is a low-grade spindle cell sarcoma with or without features resembling an endometrial stromal sarcoma or fibrosarcoma.19, 20, 21 Heterologous elements are present in 20–25% of adenosarcomas and include non-sarcomatous striated muscle, cartilage and fat; these cases are the diagnostic dilemmas between adenosarcoma and embryonal rhabdomyosarcoma.22 An important qualification is that the embryonal rhabdomyosarcoma pattern seen in an adenosarcoma is a manifestation of sarcomatous overgrowth in contrast to an exclusive pattern of embryonal rhabdomyosarcoma as in the 14 cases in the current series and the 13 cases of Daya and Scully.7
Some of the difficulties in the distinction between adenosarcoma and embryonal rhabdomyosarcoma are illustrated in two published cases of ‘adenosarcoma’ of the cervix, both presenting in 15-year-old patients.8, 23 Duggal et al23 reported the case of a 15-year-old female patient with a cervical neoplasm composed of benign endocervical glands with a ‘low-grade stromal sarcoma’ and a ‘high-grade sarcoma’ constituting 70% of the tumor area; as described, this tumor is the archetype of an adenosarcoma with sarcomatous overgrowth. By contrast, the second case, also a 15-year-old female patient, presented with a friable cervical mass with benign glands, cartilage and embryonal rhabdomyosarcoma with features of sarcoma botryoides.8 This neoplasm is an embryonal rhabdomyosarcoma of the cervix with heterologous cartilage and unlikely to be an adenosarcoma as reported.
The distinction between embryonal rhabdomyosarcoma and adenosarcoma of the uterine cervix is not a clinically inconsequential one because of therapeutic differences and syndromic manifestations. Whereas a conservative surgical management is the recommended approach in non-metastatic embryonal rhabdomyosarcoma of the female genital tract, including the cervix in children and adolescents, adenosarcoma of the cervix is more likely than not to necessitate a radical hysterectomy (with some exceptions in younger patients to preserve fertility) and, in the presence of sarcomatous overgrowth, even bilateral salpingo-oophorectomy.21, 24, 25, 26, 27, 28, 29 The relatively non-aggressive behavior of localized embryonal rhabdomyosarcomas of the cervix was initially appreciated by Daya and Scully.7 In 12 of the 14 cases in our series, local excision of the polyp followed by multidrug chemotherapy and a post-therapy biopsy or local excision alone comprised the management plan (Table 1). One patient developed a local recurrence, had a subsequent radical hysterectomy and remains tumor-free 18 years later (Case 2). The 15-year-old patient who presented with a mass rather than a polyp had chemotherapy, followed by a radical hysterectomy (Case 8). Another patient, the 32-year-old woman (Case 14) chose local excision alone and regular follow-up examinations with cervico-vaginal cytologies and has remained tumor-free 6 years after the diagnosis. However, the relatively rare occurrence of these tumors in the cervix has limited the evaluation of biological behavior and optimal medical and surgical therapy.
Although adenosarcoma with heterologous elements and/or sarcomatous overgrowth is the other main diagnostic consideration in cases of embryonal rhabdomyosarcoma of the cervix, several other rare tumors may also be in the differential diagnosis. As mentioned above, a malignant mixed Mullerian tumor or carcinosarcoma can grow in an exophytic manner from the uterine wall or cervix and have a grossly and microscopically sarcomatous appearance; however, unlike, embryonal rhabdomyosarcoma, malignant mixed Mullerian tumor typically occurs in older patients, is associated with a poor prognosis and by definition has a malignant epithelial component in addition to the malignant mesenchymal component.30 In fact, the outcome is related to the histological type and grade of the carcinoma, and metastases are usually epithelial.31 Low-grade stromal sarcoma, similar to that occurring in the uterine corpus, can rarely originate in the cervix and is distinguished by its intramural (typically not polypoid) growth, spindled stromal cells with condensation around vessels (not glands), negative immunostaining for desmin and pathognomonic chromosomal translocation (JAZF1-JJAZ1 fusion gene).32 Even a benign endocervical polyp, like the benign fibroepithelial stromal polyp, can elicit concern for an aggressive tumor, but typically lack the periglandular cuffing, phyllodes-like glandular architecture, cytological atypia of stromal cells, except in a small subset of cases and high mitotic rate seen in adenosarcoma.33
Three cases in our study had another extracervical neoplasm with provocative syndromic implications (Cases 4, 11 and 12). Less than a decade after the initial report of pleuropulmonary blastoma, Priest et al34 reported that pleuropulmonary blastoma may be a marker neoplasm for a familial tumor predisposition syndrome in as many as 40% of cases. The other tumors in addition to the pleuropulmonary blastoma include cystic nephroma of the kidney, embryonal rhabdomyosarcoma in extrapulmonary sites, nasal chondromesenchymal hamartoma, ciliary body medulloepithelioma, sex-cord stromal tumors of the ovary, rhabdomyosarcoma in other sites, and nodular hyperplasia and papillary carcinoma of the thyroid.35, 36 A germline mutation in DICER1 has recently been identified in a linkage analysis of four families with an inherited predisposition to pleuropulmonary blastoma.37 A multiloculated cyst discovered in the right upper lung lobe of one of our patients (Case 11) during preoperative staging was an involuted or ‘regressed’ pleuropulmonary blastoma.38 Indeed, there were cystic areas in her cervical embryonal rhabdomyosarcoma with an uncanny resemblance to the involuted or regressed pleuropulmonary blastoma. After the appropriate enrollment, genetic testing was performed on this patient and her family, and identified a DICER1 germline mutation she had inherited from her mother. The second case (Case 12) was the presentation of a type III pleuropulmonary blastoma at 3.5 years of age, with a disease-free interval of almost 9 years when this patient subsequently presented with a botryoid embryonal rhabdomyosarcoma of the cervix. The DICER1 status is currently under investigation in this patient. Daya and Scully7 reported the occurrence of Sertoli–Leydig cell tumors in two of the 13 cases of cervical rhabdomyosarcoma and it was the presence of masculinizing signs from such a tumor that brought the cervical embryonal rhabdomyosarcoma in Case 4 to clinical attention as an incidental finding in the gynecological examination. An enlarged thyroid gland was also detected in this same patient and a subsequent thyroidectomy revealed multinodular hyperplasia. At last contact, this patient had developed a pelvic recurrence of the Sertoli–Leydig cell tumor. Although we have no available family history of pleuropulmonary blastomas or associated tumors in Case 4, the concurrent neoplasms of the cervix and ovary and the nodular hyperplasia of the thyroid are likely manifestations of the familial pleuropulmonary blastoma tumor predisposition and dysplasia syndrome.34, 36 There are at least three additional cases in the literature of the coincidence of Sertoli–Leydig cell tumor and rhabdomyosarcoma of the cervix.39, 40, 41
Rhabdomyosarcomas in children occur as sporadic neoplasms in 90% or more of cases, but can be associated with the Li–Fraumeni syndrome, neurofibromatosis 1, and the recently recognized constitutional mismatch repair-deficiency syndrome.42, 43, 44, 45 It has been estimated that as many as 5–9% of children with a rhabdomyosarcoma have a germline TP53 gene mutation.46 It is not entirely surprising that rhabdomyosarcoma, exclusively of the botryoid or non-botryoid embryonal type, is one of the associated neoplasms in the familial pleuropulmonary blastoma tumor predisposition and dysplasia syndrome since virtually all pleuropulmonary blastomas in their initial cystic or type I stage demonstrate the morphological and immunophenotypic features of embryonal rhabdomyosarcoma.38 The cambium layer with its relationship to the overlying epithelium reflects the perturbation or disruption in the normal epithelial–stromal interaction as a consequence of a loss-of-function mutation of DICER1.32
This report not only documents our experience with the highly uncommon, cervical rhabdomyosarcoma presenting before the age of 20 years, but also highlights its possible syndromic association. As first noted by Daya and Scully,7 these tumors can often be managed in a more conservative manner, with a favorable outcome in most cases. A pathological distinction should be drawn between cervical embryonal rhabdomyosarcoma, adenosarcoma with heterologous elements and other mimics because these tumors are often managed differently and may be associated with different clinical sequelae. Finally, rhabdomyosarcoma in children should be viewed and managed in a broader context to include the possibility of familial pleuropulmonary blastoma tumor predisposition syndrome. We are unable to conclude at this point whether cervical embryonal rhabdomyosarcoma has a greater likelihood of association with familial pleuropulmonary blastoma tumor predisposition and dysplasia syndrome than does rhabdomyosarcoma arising in other anatomic sites. Likewise, we do not know whether any of the other patients in our study had the latter syndrome or one of the other rhabdomyosarcoma-associated familial syndromes.
References
Ognjanovic S, Linabery AM, Charbonneau B, et al. Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975–2005. Cancer 2009;115:4218–4226.
Dehner LP . Soft tissues. In: Stocker TJ, Dehner LP, Husain A (eds). Stocker and Dehner's Pediatric Pathology, 3rd edn. Wolter Kluwer–Lippincott Williams & Wilkins: Philadelphia, 2011, pp 1079–1087.
Leuschner I, Harms D, Mattke A, et al. Rhabdomyosarcoma of the urinary bladder and vagina. A clinicopathologic study with emphasis on recurrent disease: a report from the Kiel Pediatric Tumor Registry and the German CWS Study. Am J Surg Pathol 2001;25:856–864.
Walterhouse DO, Meza JL, Breneman JC, et al. Local control and outcome in children with localized vaginal rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children's Oncology Group. Pediatr Blood Cancer 2011;57:76–83.
Herzog CE . Overview of sarcomas in the adolescent and young adult population. J Pediatr Hematol Oncol 2005;27:215–218.
Soliman H, Ferrari A, Thomas D . Sarcoma in the young adult population: an international view. Semin Oncol 2009;36:227–236.
Daya DA, Scully RE . Sarcoma botryoides of the uterine cervix in young women: a clinicopathological study of 13 cases. Gynecol Oncol 1988;29:290–304.
Bagga R, Keepanasseril A, Srinivasan R, et al. Adenosarcoma of the uterine cervix with heterologous elements: a case report and review of literature. Arch Gynecol Obstet 2010;281:669–675.
Online Mendelian Inheritance in Man, OMIM (TM) Johns Hopkins University, Baltimore, MD. MIN Number: 601200:{9/9/2009}. World Wide Web URI: http://www.ucloi.nik.gov/omin.
Wexler LH, Meyer WH, Helman LJ . Rhabdomyosarcoma. In: Pizzo PA, Poplack DG (eds). Principles and Practice of Pediatric Oncology, 6th edn. Wolter Kluwer–Lippincott Williams & Wilkins: Philadelphia, 2011, pp 923–953.
Villela JA, Bogner PN, Jani-Sait SN, et al. Rhabdomyosarcoma of the cervix in sisters with review of the literature. Gynecol Oncol 2005;99:742–748.
Koukourakis GV, Kouloulias V, Zacharias G, et al. Embryonal rhabdomyosarcoma of the uterine cervix. Clin Transl Oncol 2009;11:399–402.
Perrone T, Carson LF, Dehner LP . Rhabdomyosarcoma with heterologous cartilage of the uterine cervix: a clinicopathologic and immunohistochemical study of an aggressive neoplasm in a young female. Med Pediatr Oncol 1990;18:72–76.
Allende DS, Yang B . Primary ovarian rhabdomyosarcoma with heterologous elements: a case report. Int J Gynecol Pathol 2008;27:402–406.
Floris G, Debiec-Rychter M, Wozniak A, et al. Malignant ectomesenchymoma: genetic profile reflects rhabdomyosarcomatous differentiation. Diagn Mol Pathol 2007;16:243–248.
Oppenheimer O, Athanasian E, Meyers P, et al. Malignant ectomesenchymoma in the wrist of a child: case report and review of the literature. Int J Surg Pathol 2005;13:113–116.
Malagon HD, Valdez Am, Moran CA, et al. Germ cell tumors with sarcomatous components: a clinicopathologic and immunohistochemical study of 46 cases. Am J Surg Pathol 2007;31:1356–1362.
Carcangu ML . Mesenchymal tumours. In: Tavassoli FA, Deville P (eds). World Health Organization Classification of Tumours, Pathology and Genetics of Tumours of the Breast and Female Genital Organs. IARC Press: Lyon, 2003, pp 280–283.
McCluggage WG, Kubik-Huch RA . Mixed epithelial and mesenchymal tumours. In: Tavalloli FA, Deville P (eds). World Health Organization Classification of Tumours, Pathology and Genetics of Tumours of the Breast and Female Genital Organs. IARC Press: Lyon, 2003, pp 284–286.
McCluggage WG . Mullerian adenosarcoma of the female genital tract. Adv Anat Pathol 2010;17:122–129.
Manoharan M, Noor Azmi MA, Soosay G, et al. Mullerian adenosarcoma of uterine cervix: report of three cases and review of literature. Gynecol Oncol 2007;105:256–260.
Clement PB, Scully RE . Mullerian adenosarcoma of the uterus: a clinicopathologic analysis of 100 cases with a review of the literature. Hum Pathol 1990;21:263–281.
Duggal R, Nijhawan R, Aggarwal N, et al. Mullerian adenosarcoma (heterologous) of the cervix with sarcomatous overgrowth: a case report with review of literature. J Gynecol Oncol 2010;21:125–128.
Martelli H, Oberlin O, Rey A, et al. Conservative treatment of girls with nonmetastatic rhabdomyosarcoma of the genital tract: a report from the Study Committee of the International Society of Pediatric Oncology. J Clin Oncol 1999;17:2117–2122.
Bernal KL, Fahmy L, Remmenga S, et al. Embryonal rhabdomyosarcoma (sarcoma botryoides) of the cervix presenting as a cervical polyp treated with fertility-sparing surgery and adjuvant chemotherapy. Gynecol Oncol 2004;95:243–246.
Gruessner SE, Omwandho CO, Dreyer T, et al. Management of stage I cervical sarcoma botryoides in childhood and adolescence. Eur J Pediatr 2004;163:452–456.
Kayton ML, Wexler LH, Lewin SN, et al. Pediatric radical abdominal trachelectomy for anaplastic embryonal rhabdomyosarcoma of the uterine cervix: an alternative to radical hysterectomy. J Pediatr Surg 2009;44:862–867.
Fleming NA, Hopkins L, de Nanassy J, et al. Mullerian adenosarcoma of the cervix in a 10-year-old girl: case report and review of the literature. J Pediatr Adolesc Gynecol 2009;22:e45–e51.
Patrelli TS, Gizzo S, Di Gangi S, et al. Cervical Mullerian adenosarcoma with heterologous sarcomatous overgrowth: a fourth case and review of the literature. BMC Cancer 2011;11:236.
Clement PB, Zubovits JT, Young RH, et al. Malignant Mullerian mixed tumors of the uterine cervix: a report of nine cases of a neoplasm with morphology often different from its counterpart in the corpus. Int J Gynecol Pathol 1998;17:211–222.
de Jong RA, Nijman HW, Wijbrandi TF, et al. Molecular markers and clinical behavior of uterine carcinosarcomas: focus on the epithelial tumor component. Mod Pathol 2011;24:1368–1379.
Boardman CH, Webb MJ, Jefferies JA . Low-grade endometrial stromal sarcoma of the ectocervix after therapy for breast cancer. Gynecol Oncol 2000;79:120–123.
Nucci MR, Young RH, Fletcher CD . Cellular pseudosarcomatous fibroepithelial stromal polyps of the lower female genital tract: an underrecognized lesion often misdiagnosed as sarcoma. Am J Surg Pathol 2000;24:231–240.
Priest JR, Watterson J, Strong L, et al. Pleuropulmonary blastoma: a marker for familial disease. J Pediatr 1996;128:220–224.
Slade I, Bacchelli C, Davies H, et al. DICER 1 syndrome: clarifying the diagnosis, clinical features and management implications of pleiotropic tumour predisposition syndrome. J Med Genet 2011;48:273–278.
Schultz KA, Pacheco MC, Yang J, et al. Ovarian sex cord stromal tumors, pleuropulmonary blastoma and DICER 1 mutations: a report from the International Pleuropulmonary Blastoma Registry. Gynecol Oncol 2011;122:246–250.
Hill DA, Ivanovich J, Priest JR, et al. DICER 1 mutations in familial pleuropulmonary blastoma. Science 2009;325:965.
Hill DA, Jarzembowski JA, Priest JR, et al. Type I pleuropulmonary blastoma: pathology and biology study of 51 cases from the International Pleuropulmonary Blastoma Registry. Am J Surg Pathol 2008;32:282–295.
Golbang P, Khan A, Scurry J, et al. Cervical sarcoma botryoides and ovarian Sertoli–Leydig cell tumor. Gynecol Oncol 1997;67:102–106.
Panagiotou JP, Polychronopoulou S, Sofou K, et al. Second and third malignant solid tumor in a girl with ovarian Sertoli–Leydig tumor. Pediatr Blood Cancer 2006;46:654–656.
McClean GE, Kurian S, Walter N, et al. Cervical embryonal rhabdomyosarcoma and ovarian Sertoli–Leydig cell tumour: a more than coincidental association of two rare neoplasms? J Clin Pathol 2007;60:326–328.
Trahair T, Andrews L, Cohn RJ . Recognition of Li–Fraumeni syndrome at diagnosis of a locally advanced extremity rhabdomyosarcoma. Pediatr Blood Cancer 2007;48:345–348.
Khayat CM, Johnston DL . Rhabdomyosarcoma, osteosarcoma, and adrenocortical carcinoma in a child with a germline p53 mutation. Pediatr Blood Cancer 2004;43:683–686.
Ferrari A, Bisogno G, Macaluso A, et al. Soft-tissue sarcomas in children and adolescents with neurofibromatosis type 1. Cancer 2007;109:1406–1412.
Kratz CP, Holter S, Etzler J, et al. Rhabdomyosarcoma in patients with constitutional mismatch-repair-deficiency syndrome. J Med Genet 2009;46:418–420.
Diller L, Sexsmith E, Gottlieb A, et al. Germline p53 mutations are frequently detected in young children with rhabdomyosarcoma. J Clin Invest 1995;95:1606–1611.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Dehner, L., Jarzembowski, J. & Hill, D. Embryonal rhabdomyosarcoma of the uterine cervix: a report of 14 cases and a discussion of its unusual clinicopathological associations. Mod Pathol 25, 602–614 (2012). https://doi.org/10.1038/modpathol.2011.185
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2011.185
Keywords
This article is cited by
-
Adolescent Embryonal Rhabdomyosarcoma of the Uterus–A Case report and Systematic Review
Indian Journal of Gynecologic Oncology (2023)
-
DICER1 tumor predisposition syndrome: an evolving story initiated with the pleuropulmonary blastoma
Modern Pathology (2022)
-
Rare Mesenchymal Tumors of Cervix: A Report of Three Cases
Indian Journal of Gynecologic Oncology (2022)
-
Embryonal rhabdomyosarcoma of the uterine corpus: a clinicopathological and molecular analysis of 21 cases highlighting a frequent association with DICER1 mutations
Modern Pathology (2021)
-
Reply: pleuropulmonary blastoma-like peritoneal sarcoma and DICER1-associated sarcomas: toward a unified nomenclature
Modern Pathology (2021)
