
Abstract
Superficial primitive Ewing's sarcomas are rare and have been reported to be of favorable prognosis compared to conventional deep-seated tumors. In the skin and subcutis, the diagnosis is often difficult, and performing molecular cytogenetic techniques may be helpful. We performed a retrospective analysis of 14 cases of superficial Ewing's sarcomas, all confirmed by molecular cytogenetics. Clinical, histological, immunohistochemical, molecular cytogenetic, therapeutic, and follow-up data are reported. There were 11 female and 3 male patients aged from 12 to 77 years (median: 17 years). Seven tumors occurred in the extremities, five in the trunk wall, and two in the head. Tumor size ranged from 1 to 5 cm (median, 3 cm). They were all small round-cell proliferations with a strong membranous positivity for CD99. Ewing's sarcoma translocations/fusion gene transcripts were detected in eight cases, both by FISH and reverse transcriptase (RT)–PCR. Four tumors were positive by RT–PCR alone (FISH not done in three cases and not interpretable in one case), and two cases were positive by FISH alone (RT–PCR not done). Surgical resection was performed in all patients. Chemotherapy was given in ten patients and radiotherapy in six. At last medical examination (median follow-up, 47 months), two patients who underwent surgical resection alone had died from the tumor. Our results confirm that superficial Ewing's sarcomas are of good prognosis. Given the difficulty of the diagnosis and the importance of an adapted treatment, a confirmation of the diagnosis by molecular or cytogenetic techniques is recommended when dealing with a superficial tumor.
Similar content being viewed by others

Main
Superficial (cutaneous/subcutaneous) malignant small round-cell tumors are relatively uncommon and include primary lesions of adnexal, melanocytic, neuroendocrine or lymphoproliferative origin, or metastatic spread from small cell carcinoma, melanoma, neuroblastoma, or rhabdomyosarcoma. Primitive Ewing's sarcoma or peripheral neuroectodermal tumors (ES/PNETs), although very rare in this location, must also be considered and have been reported as single cases1, 2, 3, 4, 5 or small series.6, 7, 8
In this unusual location, diagnosis is difficult given the poor specificity of histological and immunohistochemical profiles9 so that the specific translocation involving the EWSR1 gene often needs to be shown. ES/PNET is a family of tumors defined by a specific translocation involving the EWSR1 gene on chromosome 22q12 which is fused to an ETS gene family, most often to the FLI-1 gene on chromosome 11q24 or to the ERG gene on chromosome 21q22 and rarely to another ETS gene family.10 This specific genetic abnormality can be routinely demonstrated by reverse transcriptase (RT)–PCR or by FISH.10, 11
The prognosis of bone and deep-seated soft tissue ES/PNET is poor despite various combinations of surgery, chemotherapy, and/or radiotherapy. In two small series,7, 8 it was suggested that superficial ES/PNET might have a more favorable prognosis. However, only 2 of the 14 cases with available follow-up were confirmed molecularly in these series.
In the present study, we examined the clinicopathologic, immunohistochemical, molecular, and follow-up characteristics of 14 new cases of superficial ES/PNET. We found that patients with superficial ES/PNET do significantly better than those with deep-seated tumor.
Materials and methods
Local ethical guidelines were followed for this study and informed consent was obtained.
Case Selection
Cases coded as superficial ES/PNET were retrieved from the pathology departments at Institut Gustave Roussy, Villejuif; Institut Bergonié, Bordeaux, Centre Léon Bérard, Lyon, France, and the Institute of Pathology, Lausanne, Switzerland.
Only primary cutaneous and/or subcutaneous tumors with no muscle or bone involvement were selected.
The diagnosis of ES/PNET was based on histology, immunohistochemistry (with a strong membranous positivity for CD99), and molecular cytogenetics (FISH and/or RT–PCR). Differential diagnoses (carcinoma, melanoma, lymphoma, Merkel cell carcinoma, poorly differentiated synovial sarcoma, and rhabdomyosarcoma) were ruled out on the basis of morphology and immunohistochemical staining. Only cases with available clinical data and follow-up were retained for the study.
A total of 31 cases were coded as superficial ES/PNET. Paraffin blocks, clinical data, and/or follow-ups were missing in 10 cases; RT–PCR and/or FISH analyses were negative in 7 cases. The remaining 14 cases were retained for the study.
Clinical data, treatment, and follow-up information were obtained from the referring pathologists (see Acknowledgements).
Immunohistochemistry
Sections (4-μm thick) were prepared from formalin-fixed, paraffin-embedded tissue blocks and stained with hematoxylin, eosin, and safranin. Immunostaining was performed in all cases with the following antibodies: CD99 (monoclonal 12E7, dilution 1:5; Dakopatts, Denmark), cytokeratin (clone CK22, monoclonal, dilution 1:20; Biodema, USA), epithelial membrane antigen (monoclonal E29, dilution 1:100; Dakopatts), desmin (monoclonal D33, dilution 1:50; Dakopatts), myogenin (monoclonal Myf4, dilution 1:20; Tebu, UK), α-smooth muscle actin (monoclonal 1A4, dilution 1:200; Dakopatts), h-caldesmon (monoclonal h-CD, dilution 1:50; Dakopatts), CD45 (monoclonal 2B11-PD7/26, dilution 1:200; Dakopatts), CD3 (polyclonal, dilution 1:100; Dakopatts), CD20 (monoclonal L26, dilution 1:100; Dakopatts), CD34 (monoclonal Qbend-10, dilution 1:30; Novocastra Laboratories Inc., Newcastle upon Tyne, UK), S100 protein (polyclonal, dilution 1:500, Dakopatts), chromogranin A (monoclonal OAK-A3, dilution 1:200; Dakopatts), synaptophysin (polyclonal, dilution 1:50; Dakopatts), and FLI-1 (monoclonal G146-222, dilution 1:200; Becton Dickinson, San Diego, CA, USA). Staining was performed using the LSAB kit (Dakopatts) with an automated immunostainer. Heat-induced epitope retrieval was used for all antibodies except for α-smooth muscle actin and S100 protein. All steps were performed at room temperature and diaminobenzidine was used as chromogen. Appropriate positive and negative controls were used throughout.
RT–PCR Analysis
Design of the multiplex RT–PCR approach for the detection of EWSR1/FLI-1 and EWSR1/ERG fusion transcripts: the diversity of the breakpoint position in the EWSR1, ERG, and FLI-1 genes leads to a wide variability of the types of fusion transcripts observed in Ewing's tumors. It is well known that primers generating long amplicons compared with short ones do not lead to significant amplification efficiency when RNA is extracted from paraffin-embedded fixed tissues. To maximize success in the amplification of degraded RNA, we amplified short sequences (80–300 bp). Taking advantage of multiplexing primers, we set up a test that can detect almost all the EWSR1/FLI-1 and EWSR1/ERG fusion transcripts observed in ES in RNA extracted from archival paraffin-embedded fixed tissues. In three multiplex PCR reactions, this assay tests for more than 15 fusion gene variants, as well as the sufficient quality of each RNA sample. For EWSR1/FLI-1 transcripts, two separate RT–PCR reactions were performed containing either the primers EWSR1-7A, FLI-1-6B, and FLI1-9B or EWSR1-6A, EWSR1-9A, FLI1-6B, and FLI1-9B (Table 1). Similarly, for EWSR1/ERG, a single RT–PCR reaction was performed containing the primers EWSR1-7A, EWSR1-9A, ERG-6B, and ERG-9B (Table 1). To guarantee the quality of every RNA sample, we always co-amplified β-actin transcripts as a positive control. Translocations and resulting fusion transcript amplicons tested for in the multiplex RT–PCR assay are shown in Tables 2 and 3.
In a retrospective analysis of 65 paraffin-embedded small round-cell tumors including 30 molecularly confirmed (ie, translocation-positive) ES and 35 non-Ewing's neoplasms, the sensitivity and specificity of this multiplex RT–PCR assay in detecting EWSR1/FLI-1 and EWSR1/ERG fusion transcripts were 95 and 100%, respectively (unpublished data).
Extraction of RNA from paraffin-embedded tissues
Total RNA was extracted from formalin-fixed, paraffin-embedded fixed tissue in 10 cases, and from snap-frozen tumor tissue in 2 cases. In brief, approximately 50 mg of tumor was removed from paraffin blocks by scraping. After deparaffinization, the pellets were suspended in 200 μl of lysis buffer (20 mmol/l Tris-HCl, pH 8.0; 20 mmol/l EDTA; 1% sodium dodecyl sulfate, and 350–500 μg proteinase K) and incubation was performed at 55°C for 16–48 h. Then, 750 μl of TRIzol-LS reagent (Invitrogen, Carlsbad, CA, USA) was added to the samples and total RNA was extracted according to the manufacturer's instructions. The RNA pellet was resuspended in 40 μl of RNase-free water and stored at −80°C.
Detection of EWSR1/FLI-1 and EWSR1/ERG fusion transcripts
To detect various types of EWSR1/FLI-1 and EWSR1/ERG fusion transcripts and to achieve maximal sensitivity, we designed three parallel multiplex one-tube RT–PCRs. Each of the master solutions contains several primers (see Tables 1, 2 and 3) that are specific for particular fusion transcripts and a pair of control primers that amplifies the ubiquitously expressed β-actin gene (see above). Primers were designed to be used in identical conditions for all RT–PCR reactions and to distinguish the various fusion types by the size of PCR products. Each multiplex RT–PCR was a single-step combined RT–PCR amplification, performed using the ThermoScript One-Step RT–PCR System (Invitrogen). RT–PCR reactions (20 μl) contained 100–200 ng of RNA extract and 0.5 μM of each of the translocation-specific primers (only 0.1 μM for the β-actin primers). Reverse transcription was done at 55°C for 30 min. After inactivation of the RT at 95°C for 5 min, 40 cycles of PCR amplification were performed at 95°C for 30 s, 62°C for 45 s, and 72°C for 75 s. RT–PCR reactions were performed in duplicate with single negative and positive samples. The reaction products were subjected to electrophoresis in 2% agarose gel and visualized by ethidium bromide staining. The assay was considered satisfactory for interpretation when the internal β-actin control 274-bp PCR product was present, the external fusion-gene positive control was positive, and the water control showed no amplified product. Sequencing of the PCR products was performed using the PerkinElmer Big-Dye Terminator cycle-sequencing kit on an ABI Prism 3700 automated sequencer (PerkinElmer).
FISH
FISH assay was performed using the Histology FISH accessory kit (Dakopatts) and commercial probes (Vysis Inc., Downer Grove, IL, USA ) according to the manufacturers' instructions. Briefly, sections from representative blocks were cut onto positively charged slides at 5 μm thickness. Deparaffinization of the sections was carried out with three 10-min immersions in Xylen, followed by two 3-min immersions in wash buffer. DNA retrieval was achieved by immersing the slides in 2-[N-morpholino]ethanesulphonic acid buffer for 10 min at 97°C, then by slowly cooling to room temperature. Slides were then subjected to pepsin digestion for 7 min at 37°C, dehydrated, and air-dried. For the FISH break-apart strategy, the commercial EWSR1 dual-color break-apart probe was used (Vysis Inc.). It combines a 500 kb Spectrum Orange-labeled probe on the centromeric side of the EWSR1 breakpoint region with an 1100 kb Spectrum Green-labeled probe localizing just telomeric to this region. Co-denaturation and hybridization were achieved by placing the slides into the Hybridizer (Dakopatts). The next day, slides were washed in stringent buffer 10 min at 65°C and nuclei were counterstained by using DAPI/antifade solution. Green and orange fluorescent signals were counted in regions of cellular tumor under a Nikon Eclipse 80i fluorescent microscope with appropriate filters, and pictures were captured using a Hamamatsu C4742-95 CCD camera and analyzed with the Genikon software (Alphelys, France). For each case, 100 nuclei were counted and a case was declared positive when at least 30% of tumor cells displayed one fusion, one green and one orange signal pattern.
Results
Clinical Features (Table 4)
There were 11 female and 3 male patients. Patient age ranged from 12 to 77 years (median, 17 years). Nine patients were younger than 20, and five older than 45. Seven cases occurred in the extremities, five in the trunk wall and two in the head. The lesions were present for 1–15 months (median, 6 months). Tumor size ranged from 1 to 5 cm (median, 3 cm). Every tumor was classified as stage 2 (T1a, N0, M0, high grade) according to the AJCC Cancer Staging Manual12 at initial diagnosis (pretreatment).
Pathological Features
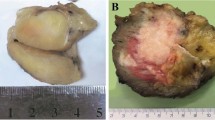
Two cases were restricted to the dermis, two were located in both the dermis and hypodermis, and ten were entirely in the hypodermis. Of the 14 cases, 7 were well demarcated (Figure 1). Two tumors were hemorrhagic and two others showed an angiomatous aspect characterized by the presence of dilated hemorrhagic spaces (Figure 2). Tumors were composed of sheets and lobules of small- to medium-sized round blue cells showing relatively regular nuclei and inconspicuous clear-to-eosinophilic cytoplasm (Figure 3). Mitotic activity ranged from 8 to 60 mitoses per 10 high power fields (median, 18 mitoses). Areas of tumor necrosis were present in nine cases, including six cases displaying less than 10% necrosis.

Gross aspect showing a well-demarcated nodule developed in the subcutis with numerous hemorrhagic spaces (patient 13).

Histological aspect at low power demonstrating a round-cell tumor with angiomatous deep-seated aspect characterized by the presence of dilated hemorrhagic spaces (patient 13).

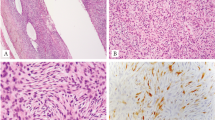
Higher magnification showing sheet of round-to-oval cells comparable to conventional/deep-seated Ewing's sarcoma (patient 13).
All tumors were strongly and diffusely positive for CD99 with a characteristic membranous staining pattern (Figure 4) and negative for muscular and lymphoid markers (Table 5). Six of seven cases were positive for FLI-1, and four of twelve contained scarce cytokeratin-positive cells. Two tumors showed focal reactivity for S100 protein, and one for neuroendocrine markers (chromogranin A and synaptophysin).

Immunohistochemistry showing a strong membranous staining for CD99 (patient 13).
Molecular Features (Table 6)
The FISH analysis showed a rearrangement of the EWSR1 gene locus in 10 cases (Figure 5). In one case, it was not possible to obtain any interpretable hybridization signals, and three tumors were not examined. Of the 12 cases examined by RT–PCR, 11 contained EWSR1/FLI-1 fusion gene transcripts, and one contained EWSR1/ERG transcripts. EWSR1/FLI-1 fusions were of type 1 (EWSR1 exon 7/FLI-1 exon 6) in 10 cases (Figure 6) and of type 2 (EWSR1 exon 7/FLI-1 exon 5) in 1 case. For the EWSR1/ERG fusion, the fusion occurred between exon 7 of EWSR1 and exon 5 of ERG.

Dual-color FISH with break-apart EWSR1 probes showing nuclei in which one pair of probe signals is split apart due to an EWSR1 region rearrangement (patient 13).

Detection of the t(11;22) (EWSR1-FLI-1) translocation using RT–PCR analysis. RT–PCR was performed with the primers necessary for amplification of the EWSR1/FLI-1 fusion gene transcripts and the ubiquitously expressed β-actin mRNA. The analysis was performed on a 2% agarose gel. M: DNA size marker (100 bp DNA ladder); 1, negative control (no RNA added); 2, positive control of the type I translocation; 3, positive control of the type II translocation; 4–5, case 13 with t(11;22) (EWSR1/FLI-1) type I translocation run in duplicate (patient 13).
Eight cases were positive with both FISH and RT–PCR, two cases were positive by FISH alone (RT–PCR not done), and four cases were positive by RT–PCR alone (FISH not done in three cases and not interpretable in one case).
Treatment and Follow-Up (Table 4)
Surgical resection of the tumor was performed in all patients. Seven patients had a wide resection whereas two had a marginal excision. Tumor excision was incomplete in four patients, and the quality of surgery not specified in one case (case 12). Ten patients received a multi-agent chemotherapy and six an adjuvant radiotherapy. By the end of the treatment, 12 patients were considered in complete remission, and 2 (cases 3 and 5) had progressive disease. Follow-up was available for 13 cases (case 12 is a recent case). The two patients with progressive disease experienced distant metastasis in bone (case 5) and in lungs (case 3) at 11 and 12 months after diagnosis and ultimately died from their tumor. The 12 other patients were alive with no evolutive disease 28–84 months (median, 47 months) after treatment completion.
Discussion
PNET and ES are malignant round-cell tumors that are regarded as two morphologic variants of the same entity, based on similar clinical, immunohistochemical and molecular profiles. The ES family10 is currently defined by the presence of specific translocations mainly involving the EWSR1 gene, which is fused to an ETS family gene (FLI-1, ERG, or ETV1). They also express CD99 in a membranous pattern, a feature which is sensitive but not specific.9 These tumors usually arise in bone and occasionally in deep soft tissues of the paraspinal region, the chest wall, and the lower extremities in children and young adults. Rarely, they occur in unusual sites, including viscera (eg, kidney, urinary bladder, prostate) and skin/subcutaneous tissue.
Since the first description of PNET/ES arising in superficial locations (ie, in the skin and/or subcutis) by Angervall and Enzinger in 1975,13 32 cases diagnosed on at least histology and CD99 positivity have been reported, more often as single cases1, 2, 3, 4, 5 with three series of 8, 5, and 13 cases.6, 7, 8 A review of the reported cases (Table 7) shows that they occurred in 8 male and 24 female patients aged from 2 to 67 years (median, 18), with no site predilection. Tumor size varied from 6 to 100 mm. The presence of ES-specific translocations was demonstrated in seven cases,1, 2, 3, 7, 8 either by classic karyotypic analysis (three cases), by RT–PCR (three cases), or FISH (three cases). A total of 30 cases were treated by surgery, 18 by chemotherapy, and 8 by radiotherapy. Of 26 patients with available follow-up, 4 died from the tumor with distant metastases. The patients who died were treated by surgery (four patients) in association with chemotherapy and radiotherapy for three patients.
In our series, there was also a female predominance (11 of 14 patients were female). The patients formed two different age groups, one between 10 and 20 years and the other over 45 years.
Owing to the rarity of the superficial location and the relative nonspecificity of its histology and immunoprofile, the diagnosis of superficial ES/PNET is often difficult and numerous differential diagnoses have to be considered in this setting. Given the importance of the treatment of ES, which should follow well-defined and validated protocols, a confirmation of the diagnosis by cytogenetics and/or molecular ancillary techniques is recommended when dealing with a superficial tumor. Demonstration of the ES fusion gene transcripts by RT–PCR performed on frozen tissue is the gold standard but a piece of tissue is rarely frozen on small superficial tumors. Thanks to recent advances in molecular biology, RT–PCR can currently be performed on formalin-fixed, paraffin-embedded tissue. FISH using EWSR1 break-apart probes is also a well-adapted method for the routine demonstration of a EWSR1 gene locus rearrangement in formalin-fixed tissue.7, 11
Thanks to multimodal therapy associating initial chemotherapy and surgery with or without radiotherapy, the prognosis of ES/PNET has significantly improved, although it still remains relatively poor with a 5-year disease-free survival of about 60% for nonmetastatic patients at presentation.14 According to the present study as well as two previously reported series,7, 8 superficial ES (ie, in the skin and/or subcutis) has a better prognosis than deep-seated lesions. In our series, the patients who died from the tumor were treated by surgery only. Conversely, the patients who were in complete remission at last follow-up had received chemotherapy (10 of 11) and radiotherapy (6 out of 11). This could suggest that superficial ES/PNETs have a good prognosis provided they are treated according to conventional ES/PNET protocols (ie, protocols for deep-seated lesions). However, in the previous reported cases, the patients who died were treated by surgery (four out of four) in association with chemotherapy and radiotherapy (three out of four).
References
Perlman EJ, Lumadue JA, Hawkins AL, et al. Primary cutaneous neuroendocrine tumors. Diagnostic use of cytogenetic and MIC2 analysis. Cancer Genet Cytogenet 1995;82:30–34.
Lee CS, Southey MC, Slater H, et al. Primary cutaneous Ewing's sarcoma/peripheral primitive neuroectodermal tumors in childhood. A molecular, cytogenetic, and immunohistochemical study. Diagn Mol Pathol 1995;4:174–181.
Sexton CW, White WL . Primary cutaneous Ewing's family sarcoma: report of a case with immunostaining for glycoprotein p30/32 mic2. Am J Dermatopathol 1996;18:601–605.
Chao TK, Chang YL, Sheen TS . Extraskeletal Ewing's sarcoma of the scalp. J Laryngol Otol 2000;114:73–75.
Somers GR, Shago M, Zielenska M, et al. Primary subcutaneous primitive neuroectodermal tumor with aggressive behaviour and an unusual karyotype: case report. Pediatr Dev Pathol 2004;7:538–545.
Banerjee SS, Agbamu DA, Eyden BP, et al. Clinicopathological characteristics of peripheral primitive neuroectodermal tumour of skin and subcutaneous tissue. Histopathology 1997;21:355–366.
Hasegawa SL, Davison JM, Rutten A, et al. Primary cutaneous Ewing's sarcoma. Immunophenotypic and molecular cytogenetic evaluation of five cases. Am J Surg Pathol 1998;22:310–318.
Ehrig T, Billings SD, Fanburg-Smith JC . Superficial primitive neuroectodermal tumor/Ewing sarcoma (PN/ES): same tumor as deep PN/ES or new entity? Ann Diagn Pathol 2007;11:153–159.
Ramani P, Rampling D, Link M . Immunohistochemical study of 12E7 in small round-cell tumours of childhood: an assessment of its sensitivity and specificity. Histopathology 1993;23:557–561.
Delattre O, Zucman J, Melot T, et al. The Ewing family of tumors—a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med 1994;331:294–299.
Yamaguchi U, Hasegawa T, Morimoto Y, et al. A practical approach to the clinical diagnosis of Ewing's sarcoma/primitive neuroectodermal tumour and other small round cell tumours sharing EWS rearrangement using new fluorescence in situ hybridisation probes for EWSR1 on formalin fixed, paraffin wax embedded tissue. J Clin Pathol 2005;58:1051–1056.
Pollock RE, Baker LH, Blumenstein B, et al. Soft tissue sarcoma. In: Greene FL, Page DL, Fleming ID, Fritz AG, Balch CM, Haller DG, Morrow M (eds). AJCC Cancer Staging Manual. Springer: New York, 2002, pp 193–197.
Angerwall L, Enzinger FM . Extraskeletal neoplasm resembling Ewing's sarcoma. Cancer 1975;36:240–251.
Oberlin O, Deley MC, Bui BN, et al., for the French Society of Paediatric Oncology. Prognostic factors in localized Ewing's tumours and peripheral neuroectodermal tumours: the third study of the French Society of Paediatric Oncology (EW88 study). Br J Cancer 2001;85:1646–1654.
Acknowledgements
We thank the following individuals for contributing cases and paraffin blocks or for supplying clinical information: Pr P Brousset, Toulouse; Dr B Chabrun, Angers; Dr C Chevreau, Toulouse; Pr S Culine, Montpellier; Dr D Cupissol, Montpellier; Dr J D'Anjou, Rouen; Dr A D'Artigues, Nîmes; Dr H Ducoin, Toulouse; Dr Durand-de-Grossouvre, Haguenau; Dr Elie, Cherbourg; Dr PP Ferrier, Albi; Dr JP Ghnassia, Strasbourg; Dr C Guillemet, Rouen; Dr JM Guinebretière, Saint-Cloud; Dr A Lecesne, Villejuif; Dr Lecigne, Cherbourg; Dr JJ Michels, Caen; Dr O Oberlin, Villejuif; Dr F Petit, Caen; Dr JM Picquenot, Rouen; Dr C Richard, Angers; Dr J Simony Lafontaine, Montpellier; Dr JL Tanné, Courseummes-sur-Mer; Dr L Zérat, Paris; Dr D Pfeiffer, Lucerne, Switzerland.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure/conflict of interest
Authors have no conflict of interest to declare.
Rights and permissions
About this article
Cite this article
Terrier-Lacombe, M., Guillou, L., Chibon, F. et al. Superficial primitive Ewing's sarcoma: a clinicopathologic and molecular cytogenetic analysis of 14 cases. Mod Pathol 22, 87–94 (2009). https://doi.org/10.1038/modpathol.2008.156
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2008.156
Keywords
This article is cited by
-
The many faces of atypical Ewing’s sarcoma. A true entity mimicking sarcomas, carcinomas and lymphomas
Virchows Archiv (2011)
-
Primitive neuroectodermal tumors in patients with testicular germ cell tumors usually resemble pediatric-type central nervous system embryonal neoplasms and lack chromosome 22 rearrangements
Modern Pathology (2010)
-
Soft tissue tumors associated with EWSR1 translocation
Virchows Archiv (2010)
