
Abstract
Once a poorly understood pathologic entity, gastrointestinal stromal tumor (GIST) has emerged in recent years as a distinct oncologic-molecular paradigm that is now a leading model for kinase-targeted therapies in oncology. Most GISTs are KIT-expressing and KIT signaling-driven mesenchymal tumors, many of which have KIT-activating mutations. A small subset of GIST show activating mutations in PDGFRA, encoding a related member of the type III receptor tyrosine kinase family. The revelation of KIT expression as a diagnostic signature of GIST has not only revolutionized the pathologic criteria in classifying GIST, but also shed light on the histogenesis of these tumors. The similarities in KIT immunoreactivity and ultrastructural appearance between GISTs and the intestinal pacemaker, the interstitial cells of Cajal (ICC), suggested that GISTs derive from or differentiate toward the ICC lineage. KIT plays a significant role in proliferation, survival, and differentiation of hematopoietic stem cells, mast cells, melanocytes, and interstitial cells of Cajal; activating KIT mutations have been identified in tumors affecting most of these cell lineages. This review will include a summary of the biology behind the specific targeted therapies, emphasizing the central role of KIT and PDGFRA oncogenic mutations in GISTs and their clinical and pathologic correlates. The role of KIT immunohistochemistry vs mutation testing will be discussed, with an insight into the indications for KIT/PDGFRA genotyping in GIST. The morphologic and molecular changes that appear with imatinib treatment, such as response and acquired imatinib resistance, are being discussed. The success GIST story based on targeted molecular paradigm may be applied in other imatinib-responsive sarcoma, such as dermatofibrosarcoma protuberans.
Similar content being viewed by others

Review of KIT and PDGFRA biology
Constitutive activation of either the KIT or PDGFRA receptor tyrosine kinase by oncogenic mutations plays a central pathogenetic role in gastrointestinal stromal tumors (GISTs).1, 2 KIT was originally identified as the cellular homolog of the retroviral oncogene v-kit in the Hardy-Zuckerman 4-feline sarcoma virus.3 In humans, the KIT gene maps to 4q12–13, in the vicinity of the genes encoding PDGFRA and FLK1 receptor tyrosine kinases. KIT belongs to class III of receptor tyrosine kinases, together with M-CSF (macrophage colony-stimulating factor) and PDGFRA, based on their sequence homology and similar conformational structure. At the N terminus, the protein contains an extracellular region made of five immunoglobulin-like repeats, followed by a single transmembrane domain including a juxtamembrane domain, and a cytoplasmic kinase domain split by a kinase insert into the adenosine triphosphate-binding and phosphotransferase regions. The only known ligand for KIT, KIT ligand (KL), was mapped on human chromosome 12q22–24. In adult life, KL is constitutively produced by some endothelial and stromal cells in the bone marrow, spleen, lymph nodes, and thymus.
The KIT receptor plays a critical role in the normal development and function of the interstitial cells of Cajal,4, 5, 6 as well as in hematopoiesis, gametogenesis, and melanogenesis during embryonic development and in the postnatal organism. Activating KIT mutations have been implicated in the pathogenesis of several other human tumors, including seminomas,7 mastocytosis,8 acute myelogenous leukemias,9 and more recently in melanomas,10 suggesting a broader role for KIT in oncogenesis.
Clinical and pathologic correlates of KIT and PDGFRA mutations
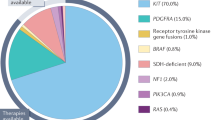
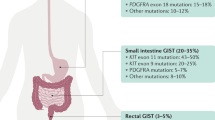
In GIST, activating KIT mutations can involve either the extracellular or the cytoplasmic domain of the receptor. The majority of the mutations (70–75%) have been found in the juxtamembrane domain, in a hotspot region at the 5′ end of exon 11, involving codons 550–560.11, 12 By analogy with other receptor tyrosine kinases, the juxtamembrane domain may function as a negative regulator of the KIT kinase and disruption of the conformational integrity of this domain may impair its negative regulatory function. Thus, the oncogenic potential of juxtamembrane domain mutations is attributed to the loss of this inhibitory function. The types of mutations occurring in this hotspot are quite heterogeneous, including in-frame deletions of variable sizes, point mutations, or deletions preceded by substitutions. A second, less common hotspot in the juxtamembrane domain is located at the 3′ end of exon 11, and these mutations are mainly internal tandem duplications (ITDs).11, 13 Activating KIT mutations in the usual exon 11 hotspot do not appear to be associated with a specific clinicopathologic phenotype, but the presence of deletions rather than substitutions predicts a more aggressive behavior.14 Specifically, deletions affecting codons 557 and 558 predict a poor prognosis.15, 16 In contrast, GIST patients harboring the above-mentioned ITDs 3′ end of exon 11 follow a more indolent clinical course and their tumors are preferentially located in the stomach.11, 13
KIT exon 9 mutations occur in 10–15% of patients and define a distinct subset of GISTs that are often located in the small bowel and show more aggressive behavior.11, 17 In contrast to the more common KIT mutations in exons 9 and 11, mutations have been rarely described in the kinase domain (exons 13 and 17).18, 19
Approximately one-third of GISTs lacking KIT mutations harbor a mutation in PDGFRA, within exon 12, 14, or 18.2, 20, 21 PDGFRA-mutated GISTs show a preference for gastric location, epithelioid morphology, variable or absent KIT expression by immunohistochemistry (IHC), and a more indolent clinical behavior.21, 22 In about 10% of patients, no detectable mutation is identified in either KIT or PDGFRA. In particular, GISTs that occur in pediatric or neurofibromatosis type I patients are nearly always wild type (ie not mutated) for both genes.23, 24, 25 In the adult population, the wild-type GIST subset represents a heterogeneous group of patients with no particular association with anatomic location or clinical outcome. In contrast, pediatric GISTs represent a distinct clinicopathologic and molecular subset, more common in females, with multifocal gastric tumors and epithelioid histology, indolent course, and absence of KIT or PDGFRA mutations.24
The role of KIT immunohistochemistry vs mutation testing and indications for KIT/PDGFRA genotyping
Although the pathologic diagnosis of GIST can be rendered on morphologic grounds in the majority of cases, as supported by KIT (CD117) immunoreactivity, in approximately 4% of cases KIT is negative by IHC.26 When compared with KIT-positive GISTs, these KIT-negative cases are more likely to have epithelioid morphology, contain PDGFRA mutations, and arise outside the gastrointestinal (GI) tract . The diagnosis of KIT-negative GISTs can be further supported by additional immunostains, which will include negativity for desmin and S-100 protein, excluding the possibility of a smooth muscle neoplasm (leiomyoma, leiomyosarcoma) or neural tumor (schwannoma). As some GISTs that are negative for KIT by IHC nonetheless contain imatinib-sensitive KIT or PDGFRA mutations, these patients should not be denied imatinib therapy based on the negative IHC result alone.
Another important point is that KIT (CD117) is not a specific marker for GIST, as weak reactivity can be seen with other mesenchymal neoplasms, such as desmoid tumors, leiomyosarcomas, and so on. Although KIT IHC remains the gold standard for GIST diagnosis, it should not replace the morphologic diagnosis and should be supplemented by other immunostains in difficult cases. In our sarcoma consultation practice, which includes cases reviewed for two multi-institutional clinical trials for adjuvant imatinib in GIST, the most commonly encountered errors were due to false-positive interpretations of KIT IHC. Approximately 5% of the GI mesenchymal tumors reviewed were erroneously interpreted as GISTs. The revised diagnosis included desmoid tumors, leiomyomas, leiomyosarcomas, spindle cell sarcoma-NOS, and so on.
An IHC KIT-negative GIST should not be confused with a KIT mutation-negative GIST (a.k.a. ‘wild-type’ GIST). This distinction is important, since wild-type GISTs are usually KIT positive by IHC, while they are the least sensitive molecular subset to imatinib therapy.
Mutational analysis is not required for all GIST patients; specifically it is not an indication in localized GISTs, which are completely resected by surgery alone. The role of adjuvant imatinib treatment is so far not well defined and ongoing clinical trails are addressing this specific question. However, the KIT/PDGFRA genotyping is critical in patients with advanced, metastatic, or recurrent disease who are undergoing therapy with selective tyrosine kinase inhibitors. Patients with KIT exon 11 mutated GIST have a very high chance (84%) of partial response with imatinib, as opposed to those with wild-type GIST, who have a partial response rate (<5%) to this drug. Furthermore, KIT exon 9 GIST might respond better to a higher dose of imatinib (800 vs 400 mg/day), and thus the genotype information might directly impact on the clinical management.27
Genotyping is also relevant to second-line treatment with sunitinib (Pfizer), as tumors with KIT exon 9 mutation or wild-type genotype have superior responses to those with KIT exon 11 mutation.
Therapy with selective kinase inhibitors: the experience with imatinib in GIST
Imatinib mesylate (STI571, Gleevec™, Novartis Pharmaceuticals, Basel, Switzerland) is a selective tyrosine kinase inhibitor whose targets include KIT and PDGFRA. Imatinib treatment achieves a partial response or stable disease in about 80% of patients with metastatic GIST.28 Recent data suggest a correlation between imatinib response and the type of mutation, as tumors with an exon 9 mutation or wild-type KIT are less likely to respond to imatinib.29, 30 Although imatinib achieves a partial response or stable disease in the majority of GIST patients, complete and lasting responses are rare. About half of the patients who initially benefit from imatinib treatment eventually develop drug resistance. The most common mechanism of acquired resistance is through a second KIT mutation, usually located in the kinase domain, which disrupts imatinib binding by stabilizing the receptor in a constitutively active form.31 Several second-generation tyrosine kinase inhibitors that show promising activity against imatinib-resistant GISTs are in the process of being tested in preclinical studies or clinical trials.
Pathologic and molecular features of imatinib response and imatinib resistance
Histologic response to imatinib can be assessed as a percentage and graded, based on the gross and microscopic areas of necrosis and fibrosis, using the following grading scheme: 1, minimal (0–10% response); 2, low (>10% and ≤50% response); 3, moderate (>50% and ≤90% response); and 4, high (>90% response).32 The histologic response of GISTs to imatinib therapy is heterogeneous, from nodule to nodule within the same resection, as well as within the same lesion. Some tumors may show only gross tumor necrosis, with large, central areas of cystification and hemorrhage, whereas the remaining solid areas are viable microscopically. Dense hyalinization with complete loss of tumor cells is rare. In most tumors showing a grade 4 response, microscopic foci of viable cells are seen either as isolated tumor cells or as distinct micronodules embedded in an extensively hyalinized background. Immunostaining for KIT may be useful in identifying and confirming residual viable tumor cells in the hyalinized areas, which could be overlooked on histologic evaluation alone. Even in tumors with a very good histologic response, small foci of distinctly viable tumor can show increased mitotic activity. The assessment of tumor proliferation in the viable foci might be a good indicator of the aggressive nature of the residual viable tumor. Thus, performing a Ki67 immunohistochemical staining, along with KIT, might be helpful in the evaluation of these responsive tumors. However, the proliferative index, assessed by Ki67 staining in the viable tumor foci, does not correlate with the degree of histologic response or duration of imatinib therapy.32
Of note, a subset of responsive GISTs may show a weaker or even negative KIT immunostaining, compared to the pre-imatinib tumor sample. Furthermore, a small group of imatinib-responsive tumors can show well-differentiated smooth muscle features, as noted by IHC and electron microscopy.32 These tumors may express desmin or other muscle markers along with weak positivity for KIT, while showing increased actin microfilaments ultrastructurally. Microarray analysis of these responsive tumors shows overexpression of genes involved in muscle development and function, when compared with that of the nontreated tumors. These findings suggest that imatinib may induce a trans-differentiation toward a smooth muscle phenotype, through chronic inactivation of KIT signaling.32 A similar phenomenon was previously described after neutralizing interstitial cells of Cajal in mice for 8 days after birth with an anti-KIT monoclonal antibody (ACK2).33
Second-site KIT mutations are rare in GISTs responsive to imatinib compared with imatinib-resistant tumors, which harbor KIT kinase domain mutations in half of the cases.31 This phenomenon seems to play a minor role in the setting of clinical response; only 1 of the 28 patients classified as clinically responsive in our study had a second-site KIT mutation.32
The structure of the KIT–imatinib complex revealed that, similar to BCR-ABL, imatinib binds the inactive conformation of the kinase, although the KIT–imatinib complex deviates somewhat from the autoinhibited inactive KIT kinase conformation. It is therefore not surprising that A-loop mutations are generally not inhibited by imatinib, although there seem to be exceptions. Therefore, there are two possible mechanisms of how resistance to imatinib therapy may develop. First, second-site mutations may stabilize the active conformation of the KIT kinase, which prevents imatinib binding. Alternatively, second-site mutations may specifically interfere with imatinib binding without affecting the overall KIT kinase conformation. Thus, most second-site mutations are located in the A-loop,31 which destabilize the inactive conformation by introducing a positively charged side chain into a positively charged pocket formed on the C-terminal lobe of the kinase. In the less common T670I, the gatekeeper residue Thr670 is replaced by an isoleucine residue. This mutation disrupts an important H-bond between imatinib and the kinase and the isoleucine methyl group protrudes into the imatinib binding site precluding proper imatinib binding to the KIT kinase.
As in CML, secondary mutations are common in imatinib resistance. The first and the second mutation are located on the same allele of the KIT gene. The second-site mutations tend to be single amino-acid substitutions in the KIT kinase domains and occur particularly in exon 17. However, secondary mutations are typically not seen in the pre-imatinib or primary resistant tumors. The mechanism for the development of a second mutation in GIST is unclear. The long duration of imatinib therapy (median of 27 months) in patients with acquired resistance who developed a second mutation makes it less likely that a pre-existing clone is responsible for acquired resistance. In contrast with CML, there is no convincing evidence of polyclonal resistance in imatinib-resistant GIST, although co-existing, multiple second mutations in different resistant nodules are reported.31 Thus, tumor nodules within a patient may develop independent means of imatinib resistance.
In half of imatinib-resistant GIST cases, there are no identifiable secondary mutations, suggesting that additional mechanisms of resistance might be responsible, such as KIT genomic amplification and activation of an alternative receptor tyrosine kinase protein in the absence of KIT expression.34
Other imatinib-responsive sarcomas: dermatofibrosarcoma protuberans
A less common mechanism of deregulating kinase signaling pathways includes translocations encoding a chimeric autocrine growth factor. One such example is the COL1A-PDGFB fusion in dermatofibrosarcoma protuberans (DFSP)35 and, more recently, the CSF1-COL6A3 fusion identified in giant cell tumor of tendon sheath.36
The recurrent t(17;22)(q22;q13) resulting in COL1A-PDGFB fusion has been reported as a consistent finding in both DFSP and giant cell fibroblastoma, supporting the concept of a common pathogenetic entity.35 The translocation fuses the strongly expressed collagen 1 alpha 1 (COL1A1) gene on chromosome 17 with the second exon of the platelet-derived growth factor-B (PDGFB) gene on chromosome 22. This distinctive translocation mechanism results in transcriptional upregulation of the PDGFB gene in the context of the COL1A1-PDGFB fusion. The post-translational processed form of the fusion protein gives rise to a fully functional and mature PDGF-B protein, which induces activation of its receptor, PDGFRB, through autocrine or paracrine routes.37 A number of clinical studies have shown a high response rate to imatinib therapy in both locally advanced and metastatic DFSP.38, 39, 40 As imatinib blocks PDGFRB signaling, these results support the concept that DFSP cells are dependent on aberrant activation of PDGFRB for cellular proliferation and survival.
Summary
The principal genetic event responsible for the pathogenesis of GIST has been identified to be a gain-of-function mutation in the KIT proto-oncogene or, occasionally, in the platelet-derived growth factor alpha (PDGFRA) gene. As these two receptor tyrosine kinases are highly sensitive targets to specific kinase inhibition when constitutively activated, GISTs have become the prototype disease for the molecular therapy of cancer. The role of pathologist has been critical in correctly identifying GISTs from other morphologically similar mesenchymal tumors of the GI tract. And although the overwhelming majority of GISTs show strong overexpression of CD117 (KIT) by IHC and can be diagnosed with certainty, 4% of tumors are negative and the correct diagnosis needs to be confirmed by KIT/PDGFRA mutation analysis. These tumors usually contain imatinib-sensitive KIT or PDGFRA mutations and thus these patients should not be denied imatinib therapy based on the negative IHC result alone. The main indications for KIT/PDGFRA mutation analysis are in the context of imatinib therapy or imatinib resistance, as a result of a direct relationship between the presence and location of KIT mutation and imatinib response.
References
Hirota S, Isozaki K, Moriyama Y, et al. Gain of function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998;279:577–580.
Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003;299:708–710.
Besmer P, Murphy JE, George PC, et al. A new acute transforming feline retrovirus and relationship of its oncogene v-kit with the protein kinase gene family. Nature 1986;320:415–421.
Huizinga JD, Thuneberg L, Kluppel M, et al. W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature 1995;373:347–349.
Maeda H, Yamagata A, Nishikawa S, et al. Requirement of c-kit for development of intestinal pacemaker system. Development 1992;116:369–375.
Torihashi S, Ward SM, Nishikawa S, et al. c-kit-dependent development of interstitial cells and electrical activity in the murine gastrointestinal tract. Cell Tissue Res 1995;280:97–111.
Tian Q, Frierson Jr HF, Krystal GW, et al. Activating c-kit gene mutations in human germ cell tumors. Am J Pathol 1999;154:1643–1647.
Nagata H, Worobec AS, Oh CK, et al. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci USA 1995;92:10560–10564.
Gari M, Goodeve A, Wilson G, et al. c-kit proto-oncogene exon 8 in-frame deletion plus insertion mutations in acute myeloid leukaemia. Br J Haematol 1999;105:894–900.
Willmore-Payne C, Holden JA, Tripp S, et al. Human malignant melanoma: detection of BRAF- and c-kit-activating mutations by high-resolution amplicon melting analysis. Hum Pathol 2005;36:486–493.
Antonescu CR, Sommer G, Sarran L, et al. Association of KIT exon 9 mutations with nongastric primary site and aggressive behavior: KIT mutation analysis and clinical correlates of 120 gastrointestinal stromal tumors. Clin Cancer Res 2003;9:3329–3337.
Rubin BP, Singer S, Tsao C, et al. KIT activation is an ubiquitous feature of gastrointestinal stromal tumors. Cancer Res 2001;61:8118–8121.
Lasota J, Dansonka-Mieszkowska A, Stachura T, et al. Gastrointestinal stromal tumors with internal tandem duplications in 3′ end of KIT juxtamembrane domain occur predominantly in stomach and generally seem to have a favorable course. Mod Pathol 2003;16:1257–1264.
Andersson J, Bumming P, Meis-Kindblom JM, et al. Gastrointestinal stromal tumors with KIT exon 11 deletions are associated with poor prognosis. Gastroenterology 2006;130:1573–1581.
Martin J, Poveda A, Llombart-Bosch A, et al. Deletions affecting codons 557–558 of the c-KIT gene indicate a poor prognosis in patients with completely resected gastrointestinal stromal tumors: a study by the Spanish Group for Sarcoma Research (GEIS). J Clin Oncol 2005;23:6190–6198.
Wardelmann E, Losen I, Hans V, et al. Deletion of Trp-557 and Lys-558 in the juxtamembrane domain of the c-kit protooncogene is associated with metastatic behavior of gastrointestinal stromal tumors. Int J Cancer 2003;106:887–895.
Lasota J, Kopczynski J, Sarlomo-Rikala M, et al. KIT 1530ins6 mutation defines a subset of predominantly malignant gastrointestinal stromal tumors of intestinal origin. Hum Pathol 2003;34:1306–1312.
Lasota J, Wozniak A, Sarlomo-Rikala M, et al. Mutations in exons 9 and 13 of KIT gene are rare events in gastrointestinal stromal tumors. A study of 200 cases. Am J Pathol 2000;157:1091–1095.
Lux ML, Rubin BP, Biase TL, et al. KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am J Pathol 2000;156:791–795.
Hirota S, Ohashi A, Nishida T, et al. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology 2003;125:660–667.
Lasota J, Dansonka-Mieszkowska A, Sobin LH, et al. A great majority of GISTs with PDGFRA mutations represent gastric tumors of low or no malignant potential. Lab Invest 2004;84:874–883.
Wardelmann E, Hrychyk A, Merkelbach-Bruse S, et al. Association of platelet-derived growth factor receptor alpha mutations with gastric primary site and epithelioid or mixed cell morphology in gastrointestinal stromal tumors. J Mol Diagn 2004;6:197–204.
Miettinen M, Lasota J, Sobin LH . Gastrointestinal stromal tumors of the stomach in children and young adults: a clinicopathologic, immunohistochemical, and molecular genetic study of 44 cases with long-term follow-up and review of the literature. Am J Surg Pathol 2005;29:1373–1381.
Prakash S, Sarran L, Socci N, et al. Gastrointestinal stromal tumors in children and young adults: a clinicopathologic, molecular, and genomic study of 15 cases and review of the literature. J Pediatr Hematol Oncol 2005;4:179–187.
Miettinen M, Fetsch JF, Sobin LH, et al. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: a clinicopathologic and molecular genetic study of 45 cases. Am J Surg Pathol 2006;30:90–96.
Medeiros F, Corless CL, Duensing A, et al. KIT-negative gastrointestinal stromal tumors: proof of concept and therapeutic implications. Am J Surg Pathol 2004;28:889–894.
Debiec-Rychter M, Sciot R, Le Cesne A, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 2006;42:1093–1103.
Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002;347:472–480.
Debiec-Rychter M, Dumez H, Judson I, et al. Use of c-KIT/PDGFRA mutational analysis to predict the clinical response to imatinib in patients with advanced gastrointestinal stromal tumours entered on phase I and II studies of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer 2004;40:689–695.
Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 2003;21:4342–4349.
Antonescu CR, Besmer P, Guo T, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res 2005;11:4182–4190.
Agaram NP, Besmer P, Wong GC, et al. Pathologic and molecular heterogeneity in imatinib-stable or imatinib-responsive gastrointestinal stromal tumors. Clin Cancer Res 2007;13:170–181.
Torihashi S, Nishi K, Tokutomi Y, et al. Blockade of kit signaling induces transdifferentiation of interstitial cells of Cajal to a smooth muscle phenotype. Gastroenterology 1999;117:140–148.
Heinrich MC, Corless CL, Blanke CH, et al. Molecular correlates of imatinib-resistance in gastrointestinal stromal tumors. J Clin Oncol 2006;24:4764–4774.
Simon MP, Pedeutour F, Sirvent N, et al. Deregulation of the platelet-derived growth factor B-chain gene via fusion with collagen gene COL1A1 in dermatofibrosarcoma protuberans and giant-cell fibroblastoma. Nat Genet 1997;15:95–98.
West RB, Rubin BP, Miller MA, et al. A landscape effect in tenosynovial giant-cell tumor from activation of CSF1 expression by a translocation in a minority of tumor cells. Proc Natl Acad Sci USA 2006;103:690–695.
Sjoblom T, Shimizu A, O'Brien KP, et al. Growth inhibition of dermatofibrosarcoma protuberans tumors by the platelet-derived growth factor receptor antagonist STI571 through induction of apoptosis. Cancer Res 2001;61:5778–5783.
Maki RG, Awan RA, Dixon RH, et al. Differential sensitivity to imatinib of 2 patients with metastatic sarcoma arising from dermatofibrosarcoma protuberans. Int J Cancer 2002;100:623–626.
Sirvent N, Maire G, Pedeutour F . Genetics of dermatofibrosarcoma protuberans family of tumors: from ring chromosomes to tyrosine kinase inhibitor treatment. Genes Chromosomes Cancer 2003;37:1–19.
McArthur GA, Demetri GD, Van Oosterom A, et al. Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: Imatinib Target Exploration Consortium Study B2225. J Clin Oncol 2005;23:866–873.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Antonescu, C. Targeted therapy of cancer: new roles for pathologists in identifying GISTs and other sarcomas. Mod Pathol 21 (Suppl 2), S31–S36 (2008). https://doi.org/10.1038/modpathol.2008.9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2008.9
Keywords
This article is cited by
-
Liver resection for metastatic GIST tumor improves survival in the era of tyrosine kinase inhibitors: a systematic review and meta-analysis
Langenbeck's Archives of Surgery (2023)
-
Endoscopic Enucleation Is Effective and Relatively Safe in Small Gastric Subepithelial Tumors Originating from Muscularis Propria
Digestive Diseases and Sciences (2019)
-
Gastrointestinal stromal tumors: Diagnosis, therapy and follow-up care in Austria
Wiener Medizinische Wochenschrift (2013)
-
Clinical practice guidelines for patients with gastrointestinal stromal tumor in Taiwan
World Journal of Surgical Oncology (2012)
-
A case of gastrointestinal stromal tumor diagnosed on prostate biopsy
Nature Clinical Practice Urology (2009)
