
Abstract
Recent studies suggest a possible link between calcification and ischemia–reperfusion injury following liver transplantation. Histological staining, immunolabeling, and biochemical and electron microscopy analyses were applied to assess the possible mechanism(s) of calcification in liver tissue. Although light microscopy studies did not reveal the presence of large necrotic or apoptotic areas, electron microscopy showed the presence of membrane-bound vacuolar structures in hepatocytes, indicative of cell damage. Myofibroblasts were abundant in regions surrounding and within calcification. In these precalcified and calcified areas, myofibroblasts expressed bone-specific matrix proteins, such as osteopontin, type 1 collagen and bone sialoprotein. In addition, transforming growth factor beta (TGFβ)-1 and BMP2, two growth factors implicated in osteoblast differentiation, and Runx2 and Msx2, two transcription factors targets of TGFβ-1 and BMP2, were also expressed in these myofibroblasts. These data suggest that liver calcification following transplantation may be a consequence of precipitation of hydroxylapatite emanating from necrotic or apoptotic hepatocytes associated with proliferation of myofibroblasts expressing bone-specific matrix proteins.
Similar content being viewed by others


Main
Cases of pathological liver calcification have been reported following a wide spectrum of injuries such as inflammatory states as well as primary benign or malignant neoplasm.1 Furthermore, clinical reports have documented diffuse hepatic calcification developing after extensive ischemia2 or metastasis.3 In all cases, calcification was a consequence of massive necrosis. In addition to necrosis, liver cell apoptosis may also predispose to calcification as previously described.4 Both necrosis and apoptosis have been shown to be massively triggered during experimental ischemia–reperfusion performed in pig liver,5 thus suggesting a potential link between ischemia–reperfusion and the observed calcifications. In spite of small patient number, calcification was found in 60% of liver disfunctions following human liver transplantation.6
Ectopic calcification is characterized by the pathological formation of hydroxyapatite crystals in nonskeletal tissues. Pathological calcifications can be subdivided into (i) ‘metastatic calcification’ occurring in undamaged tissues when calcium and phosphate serum levels are elevated and (ii) ‘dystrophic calcification’ occurring in injured tissue when the levels of calcium and phosphate in serum are normal.7 Dystrophic calcifications have many features similar to physiological bone mineralization such as the deposition of hydroxyapatite in organic matrix containing type I collagen and noncollagenous proteins. Bone mineralization is regulated by noncollagenous proteins present in bone organic matrix such as osteopontin, bone sialoprotein and osteocalcin.8 These bone-specific genes are activated during osteogenic differentiation by several growth factors such as transforming growth factor beta (TGFβ)-1 and BMP2, which are able to activate the specific transcription factors, Msx2 and Runx2.9, 10 Runx2 can cooperate with TGFβ- and BMP-specific Smads and stimulate transcription of the target genes required for osteogenesis.11, 12
We previously reported two cases of graft calcification following liver transplantation,6, 13 in which patients had developed massive dystrophic liver calcification after normal ischemia–reperfusion periods. In the present study, we have used a combined approach involving histological staining, immunolabeling, electron microscopy and biochemical analysis to assess the involvement of the necrosis–apoptosis and the osteogenesis in the process of liver calcification.
Materials and methods
Tissue Collection
Calcified liver was obtained as previously described by Tzimas et al,13 macroscopically divided into noncalcified, precalcified and calcified parts and snap frozen in less than 5 min. Three samples from each region were dissected and used for biochemical and morphological studies. Control livers were obtained from biopsies collected in the operating room in accordance with the McGill University Health Centre ethics regulations (ERB 05-003).
Histology
Hematoxylin and eosin staining and von Kossa staining were carried out following the procedure described by Miao and Scutt14 and Xue et al,15 respectively. Masson–Goldner trichrome staining was performed as previously described.16 Hematoxylin and eosin, von Kossa and Masson–Goldner trichrome were performed on paraffin-embedded sections. For Alizarin red S staining, 8 μm frozen sections were fixed with 10% formalin, partly decalcified in 10 mM ethylenediaminetetraacetic acid (EDTA, five washes of 2 min) and stained with 40 mM Alizarin red S (Sigma) for 5 min at room temperature. The sections were then washed three times with PBS and counterstained with hematoxylin (Vector Laboratories).
Immunohistochemistry
Frozen sections (8 μm) were fixed with 4% parafomaldehyde in PBS (pH 7.4) and partly decalcified with 10 mM EDTA (five washes of 2 min). Sections were incubated in 3% hydrogen peroxide to quench endogenous peroxidases and blocked with 1% Bovine Serum Albumin (BSA, Sigma) in PBS. The sections were then incubated with specific primary antibodies for 1 h at 37°C in blocking solution. Mouse monoclonal antibodies against alpha smooth muscle actin (α-SMA, clone 1A4, Dako, 1:1000), Keratin 18 (Labvision Corporation, 1:500), human CD31 (Chemicon International, 1:1000), TGFβ-1 (clone1D11, R&D Systems, 1:50) or rabbit sera against Runx2 (Santa Cruz, 1:50), Msx2 (Santa Cruz, 1:50), BMP2 (Santa Cruz, 1:50) and Ki-67 (1:200) were used. After three washes in PBS, sections were incubated with Envision amplification system Dako (Cytomation Envision+System labeled polymer-HRP anti-mouse or anti-Rabbit) for 30 min at room temperature and then washed three additional times. Sections were stained with diaminobenzidine (DAB, Dako) as chromogen and counterstained with hematoxylin. Paraffin-embedded sections (5 μm) were prepared for type I collagen (goat anti-human type I collagen antibody, Southern Biotechnology Associates), osteocalcin (goat anti-mouse osteocalcin, Biomedical Technologies), osteopontin (rabbit anti-osteopontin, LF-123) and bone sialoprotein (rabbit anti-human bone sialoprotein, LF-6) detections following procedure described.17 After overnight incubation at room temperature, incubation with specific biotinylated rabbit anti-goat IgG or goat anti-rabbit IgG (Sigma) and incubation with the Vectastain ABC-AP kit (Vector Laboratories, Inc., Ontario, Canada), red pigmentation was produced by a 10- to 15-min treatment with Fast Red TR/Naphthol AS-MX phosphate (Sigma; containing 1 mM levamisole as endogenous alkaline phosphatase inhibitor). After washing with distilled water, the sections were counterstained with methyl green and mounted with Kaiser's glycerol jelly. In all experiments, the specificity of the staining was controlled by omitting the primary antibody.
Cell Apoptosis Assay
Terminal-deoxynucleotidyl transferase-mediated nick-end labeling (TUNEL) staining of apoptotic cells was performed on 8 μm frozen sections according to the manufacturer's instruction (Roche) as described previously.18
Transmission Electron Microscopy
Approximately 0.02 cm3 tissue specimens were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer, washed with cacodylate buffer and post-fixed with 1% OsO4. The fixed material was then dehydrated in graded concentrations of alcohol, and embedded in Epon resin. Ultrathin sections were prepared by ultramicrotomy using a diamond knife. To distinguish between mineralized and nonmineralized cellular structures, lead citrate- and uranium acetate-stained and nonstained sections were imaged in transmission electron microscopy using a JEOL JEM 2000FX transmission electron microscope (Tokyo, Japan) operated at 80 kV. The chemical composition of the mineral phases was determined by energy dispersive spectroscopy (Quartz XOne).
Immunoblot Analyses
Liver tissues (1–2 g) were homogenized in 1 ml of PBS containing 1 × protease inhibitor cocktail (Roche), 5 mM NaF, 1 mM EDTA and 1 mM Na3VO4 with an Ultra-Turrax T25. Tissue remnants were removed by centrifugation at 1500 g for 20 min at 4°C. Membrane proteins were solubilized by addition of TX-100 to a 1% final concentration. After incubation on ice for 1 h, insoluble material was removed by centrifugation for 1 h at 100 000 g. Protein concentrations were determined using the Bradford reagent. Samples were immunoblotted as previously described19 using antibody dilutions as follow: 1/200 (Runx2, Msx2 and BMP2), 1:500 (osteopontin, bone sialoprotein and collagen I) and 1:1000 (TGFβ-1).
Results
Characterization of Liver Calcifications
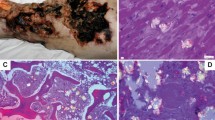
Calcified livers were collected 15 days post-transplantation as previously described.13 All tissue sections and proteins analyzed originated from these livers. The presence of calcification within the liver tissues was evident by histochemical analysis using three different methods. Von Kossa staining showed the presence of phosphate as indicated black silver precipitation within liver cells, most likely hepatocytes (Figure 1, top panels, von Kossa), whereas Alizarin Red S staining indicated the presence of calcium (Figure 1, middle panels, Alizarin Red S). The co-existence of calcium and phosphate suggests the precipitation of a calcium phosphate phase within the calcified tissue. Goldner–Masson trichrome staining (Figure 1, bottom panels, Goldner–Masson) confirmed the presence of collagen-containing matrix in the calcified areas, as indicated by the light grey precipitates.

Characterization of liver calcification. Von Kossa (top panels), alizarin red S (middle panels) and Goldner–Masson trichrome (bottom panels) staining of calcified, precalcified and noncalcified liver tissue sections. Top panels, black staining indicates the presence of phosphate precipitate. Middle panels, dark grey staining indicates the presence of Ca2+ precipitate. Bottom panels, light grey staining indicates the presence of collagen. Magnification × 20.
Tissue Necrosis and Apoptosis
Pathological calcifications have previously been reported to be associated with necrosis and apoptosis.4 To assess the presence of necrotic areas in calcified liver, we first stained liver sections with hematoxylin and eosin. Unexpectedly, necrosis was not detected in any of the sections including noncalcified (Figure 2a), precalcified (Figure 2a) or calcified tissues (not shown). Similarly, TUNEL staining showed labeling of only a small proportion of cell nuclei (Figure 2b, arrows) but did not reveal massive apoptosis in any areas of the livers (Figure 2b). On the contrary, in precalcified region of the liver, many cells were positively stained with the anti-Ki-67 antibodies, indicating their proliferative state (Figure 2c, arrows). Although light microscopy studies did not reveal massive necrosis/apoptosis, electron microscopy analysis of the precalcified sections showed the presence of membrane-bound vacuolar structures, most likely altered mitochondria in hepatocytes (Figure 3c), indicative of cell damage.20, 21 Furthermore, a significant number of intact hepatocytes within the calcified area showed the presence of intracellular calcium phosphate precipitates (Figure 3a and d), confirming the results of von Kossa staining (Figure 1). Calcium phosphate precipitates were found in the cytosol and were not associated to an organelle such as vacuole-like or mitochondria (this study and that of Tzimas et al6). Chemical analysis by energy-dispersive spectroscopy and high-resolution transmission electron microscopy imaging indicated the presence of hydroxyapatite crystals similar to those observed in bone as well as hydroxyapatite formed in atherosclerosis.22 Additional hydroxyapatite precipitates were also present in association with extracellular filamentous structures, suggesting a ‘classical’, extracellular matrix-associated calcification process (Figure 3b).

Characterization of necrotic, apoptotic and proliferative states. Hematoxylin and eosin staining (a), TUNEL staining (b) and Ki-67 immunostaining (c) of liver sections obtained from noncalcified or calcified regions. Arrows show apoptotic nuclei (b) or cells in proliferation (c). Magnification × 20 (a), × 40 (b–c). In Ki-67 control experiment, no labeling was observed.

Electron microscopy analyses. Calcified (a) and fibrotic (b) regions were analyzed by transmission electron microscopy. The presence of dilated mitochondria was detected in numerous hepatocyte-like cells (c) as well as hydroxyapatite deposits within cell cytoplasms (d). Bars indicate either 5 μm (a–c), 1 μm (b) or 2 μm (d).
Identification of Major Cell Populations in Calcified Liver
The presence of at least two major cell populations in precalcified areas was evident by hematoxylin and eosin staining (Figure 2a). To further characterize these cells, immunohistolabeling experiments were carried out on noncalcified and precalcified liver sections. Antibody against Keratin 18, a specific marker of hepatocytes, showed a predominant labeling in the parenchyma of noncalcified regions (Figure 4a; top panel). Interestingly, Keratin 18-positive hepatocytes were not present in large numbers in the area adjacent to the calcified tissue where, at least, another cell type was predominant (Figure 4a; bottom panel). Immunostaining with anti-αSMA antibodies specific for myofibroblasts and smooth muscle cells demonstrated the presence of significant numbers of immunostained cells at the periphery of blood vessels in noncalcified regions (Figure 4b; top panel), indicating the presence of smooth muscle cells. Remarkably, a large number of cells at the edge of and within calcified areas (Figure 4b; bottom panel and not shown, respectively) also expressed αSMA, suggesting the proliferation of myofibroblasts in these regions in response to liver damage.23 Finally, as endothelial cell proliferation has been reported to be associated with fibrosis,24 liver sections were labeled using an antibody against CD31, a specific endothelium marker. No differences were observed, however, in any of the sections obtained from different regions (Figure 4b). These results provide evidence for a massive proliferation of myofibroblasts in both precalcified and calcified regions.

Characterization of the different cell populations in noncalcified (top panels) and precalcified (bottom panels) regions. Immunostaining was performed with antibodies against the hepatocyte marker Keratin 18 (a), the myofibroblast marker αSMA (b) and the endothelial cell marker CD31 (c). Magnification × 40. In control experiments, no labeling was observed; moreover, all three antibodies are mouse monoclonal from the same isotype and showed different labeling.
Expression of Specific Bone Markers in Calcified Liver
Physiological and dystrophic calcification is a highly regulated process involving the synthesis and secretion of collagenous and noncollagenous proteins forming the bone matrix. To test whether bone matrix proteins played a role in liver calcification, we first examined their presence in the different regions of the calcified livers using immunolabeling using antibodies against type 1 collagen and other noncollagenous proteins such as osteopontin, osteocalcin and bone sialoprotein. Osteopontin, a secreted glycoprotein with the ability to bind to Ca2+ was expressed in myofibroblasts in both calcified and precalcified areas (Figure 5a, osteopontin). In contrast, osteocalcin, a highly abundant noncollagenous protein of bone matrix, was only faintly expressed in myofibroblasts and hepatocytes in all three regions (data not shown). The expression of type I collagen, the main organic compartment of bone but also an extracellular matrix component during fibrosis, was restricted to myofibroblasts in both precalcified and calcified regions of the livers (Figure 5a, type 1 collagen). Finally, bone sialoprotein, a promoter of osteoblast differentiation and a potential nucleator of hydroxyapatite crystals, was expressed by both hepatocytes and myofibroblasts (Figure 5a, bone sialoprotein). The results of immunohistochemistry were confirmed by immunoblot analyses (Figure 5b). Indeed, the expression of both osteopontin and type I collagen increased in precalcified and calcified regions of the liver relative to noncalcified areas. We were unable to detect bone sialoprotein by immunoblot. These results confirmed the presence of osseous extracellular matrix components that may reflect an important fibrogenic process. They also suggest that an active bone-forming process may operate simultaneously to myofibroblast proliferation.

Bone matrix protein expression in the liver. Immunostaining (a) of calcified (top panels), precalcified (middle panels) and noncalcified (bottom panels) tissue sections was carried out using antibodies against osteopontin, or type 1 collagen or bone siaoloprotein. Magnification × 20. In control experiments, no labeling was observed. In (b), Western blot analysis of calcified, precalcified and noncalcified tissue extracts were carried out using antibodies against osteopontin or type 1 collagen. Antibody against β-actin was used as a control.
TGFβ Superfamily and Liver Calcification
The results presented above suggest a correlation between the presence of collagenous and noncollagenous bone-forming matrix proteins and myofibroblast proliferation associated with the deposition of hydroxyapatite minerals. It is well established that the TGFβ superfamily represents a common denominator to both fibrosis induction and bone formation. As a consequence, we analyzed the expression and localization of two major members of the TGFβ superfamily, namely TGFβ-1 and BMP2 in noncalcified and in precalcified sections. Interestingly, while TGFβ-1 immunostaining labeled both myofibroblasts and hepatocytes (Figure 6a), BMP2 was detected predominately in myofibroblasts and in rare cases in hepatocytes (Figure 6a). These results were confirmed by immunoblot analysis on proteins extracted from the three regions of the liver. Indeed, both BMP2 and TGFβ-1 protein expression levels were increased in protein extracts from both precalcified and calcified regions (Figure 6b).

TGFβ family growth factor and osteoblastic transcription factor expression. Immunostaining (a) of precalcified (right panels) and noncalcified (left panels) liver tissue sections was carried out using antibodies raised against TGFβ-1, BMP2, Runx2 or Msx2. Magnification × 40. In control experiments, no labeling was observed; moreover, BMP2, Runx2 and Msx2 are polyclonal rabbit antibodies and they showed distinct labeling patterns, especially in noncalcified sections. In (b), Western blot analysis of calcified, precalcified and noncalcified tissue extracts were carried out using antibodies against TGFβ-1, BMP2, Msx2 or Runx2. Antibody against β-actin was used as a control.
To further confirm these observations and assess their functional significance, we evaluated the expression of specific transcription factors whose expression is regulated by TGFβ-1 or BMP2 and which have been implicated in osteoblast differentiation and calcified matrix synthesis. Two of these transcription factors Runx2 (Figure 6a) and Msx2 (Figure 6a) were selected based on their previously reported dependence on TGFβ-1 and BMP2.25 These two transcription factors were mainly immunodetected in myofibroblasts present in precalcified regions (Figure 6a), whereas in noncalcified areas only cells surrounding blood vessels were stained (Figure 6a). These results were confirmed by immunoblot analysis, with enhanced expression of Msx2 and Runx2 in protein extracts from precalcified and calcified areas (Figure 6b). These data confirm the presence of TGFβ-1 and BMP2 with their targets Runx2 and Msx2 in myofibroblasts. Although the overall calcification pattern in liver appears to be distinct, the expression of these proteins suggests that myofibroblasts may also undergo specific osteoblastic transformation leading to the synthesis of calcified matrix similar to that observed in pathological aortic calcification.25
Discussion
Patients undergoing liver transplantation occasionally develop complications such as calcifications which may in turn lead to graft dysfunction.13 Frequently, microcalcifications are detected in transplanted livers and are attributed to ischemia–reperfusion injury.2 On some occasions, however, the development of macrocalcifications has been observed, leading to graft nonfunction and the requirement of a new transplantation.13 Unfortunately, the mechanism of pathological calcification is poorly understood, particularly in the case of liver calcification. This study represents the first attempt to characterize the events leading to liver calcification obtained from partially calcified transplanted livers.
Characterization of Liver Injury
The results of von Kossa, Alizarin red S staining, transmission electron microscopy and energy-dispersive spectroscopy analyses provide clear evidence that liver calcification is due to precipitation of crystalline hydroxyapatite (Figure 1). Considering that the liver was removed 15 days after the initial transplant, no sign of liver necrosis was detectable by hematoxylin and eosin staining. In contrast, transmission electron microscopy images showed the presence of degenerating cells containing dilated mitochondria, indicative of significant cell injury (Figure 3c). It is possible that hematoxylin and eosin staining may not be sufficient to completely identify necrotic tissues. Furthermore, the paucity of TUNEL-positive cells at the periphery of calcified regions suggested that massive apoptosis may not have occurred at the initial stage of liver calcification. In a pig model of liver ischemia–reperfusion, sinusoidal cell and hepatocyte apoptosis has been shown to increase over time and to almost return to background level after 24 h of reperfusion.5 In addition, more than 35% of hepatocytes have been found to undergo necrosis.5 As our samples were collected 15 days post reperfusion, it cannot be excluded that a massive apoptosis occurred shortly after transplantation and therefore may have been at the origin of the calcification process. A large number of Ki-67-positive proliferating hepatocytes was observed (Figure 2c), which suggested that a regeneration process may have been occurred after injury as has previously been reported.26 These data are in agreement with the increased phosphorylation of ERK1/2 in proteins extracted from the precalcified and calcified regions (data not shown), suggesting a growth factor-activated regeneration process as previously described.27 In addition, the presence of abundant proliferating myofibroblasts (αSMA-positive cells), as shown by Ki-67 staining (Figure 2c), suggests the existence of mitogens in their microenvironment.
Possible Mechanisms of Calcification
Histological Von Kossa and Alizarin red S staining suggested an intracellular accumulation of calcium phosphate in damaged hepatocytes (Figure 1). Based on high-resolution transmission electron microscopy imaging and energy-dispersive spectroscopy analysis, the structure and composition of the calcium phosphate is consistent with hydroxyapatite. Morphological features characteristic for phagocytosis and endocytosis of hydroxyapatite precipitates were not observed in both myofibroblasts and hepatocytes. Transmission electron microscopy shows the presence of hydroxyapatite within fibrotic regions similar to the pattern observed in arterial calcification.28 The detection of osteopontin, osteocalcin, bone sialoprotein and collagen I, four proteins found in calcified osseous matrix, leads to the assumption that activated myofibroblasts may synthesize and secrete these proteins. Indeed, it has been shown that osteopontin produced locally by the smooth muscle cells may serve as an inducible inhibitor of vascular calcification.29
Among the growth factors implicated in the mechanisms triggering myofibroblasts-dependent calcification, TGFβ-1 and BMP2 are strong osteo-inducers.30 Moreover, TGFβ-1 is known to induce type 1 collagen production in osteoblastic cells31 and to participate in type 1 collagen synthesis by myofibroblasts and in calcium deposition in fibrotic areas. These growth factors were mainly expressed in myofibroblasts (Figure 6) of the liver samples consistent with our findings on the production of osseous matrix proteins by these cells. Msx29 and Runx210 are two transcription factors implicated in osteoblast differentiation and in arterial calcification. They are known targets of TGFβ-1 and BMP2.25 These transcription factors were highly expressed in myofibroblasts present in precalcified and calcified areas. This correlated with the expression of osteopontin, osteocalcin, bone sialoprotein and type I collagen (this study and that of Otto et al10). Taken together, these data suggest that besides the contribution of hydroxyapatite deposition within necrotic hepatocytes, myofibroblast-dependent calcification may also occur in the calcified liver by a mechanism which is dependent on TGFβ-1 and BMP2 and their downstream transcriptional targets.
In conclusion, in this study we have demonstrated that liver macrocalcification following transplantation may be the consequence of a generalized response to injury involving osteoblast-like differentiation of activated myofibroblasts. In addition, we have demonstrated the presence of intracellular accumulation of hydroxyapatite deposits in hepatocytes, which may be associated with their ischemia/reperfusion-induced injury. We believe that these two mechanisms may both contribute to the process of liver calcification and its pathological consequences. Inhibition of at least one of these mechanisms may, therefore, prevent massive liver calcification and improve organ recovery following injury.
References
Stoupis C, Taylor HM, Paley MR, et al. The Rocky liver: radiologic–pathologic correlation of calcified hepatic masses. Radiographics 1998;18:675–685; quiz 726.
Shibuya A, Unuma T, Sugimoto T, et al. Diffuse hepatic calcification as a sequela to shock liver. Gastroenterology 1985;89:196–201.
Shih WJ, Han JK, Magoun S, et al. Bone agent localization in hepatic metastases. J Nucl Med Technol 1999;27:38–40.
Kim KM . Apoptosis and calcification. Scanning Microsc 1995;9:1137–1175; discussion 1175–1138.
Meguro M, Katsuramaki T, Kimura H, et al. Apoptosis and necrosis after warm ischemia–reperfusion injury of the pig liver and their inhibition by ONO-1714. Transplantation 2003;75:703–710.
Tzimas GN, Afshar M, Emadali A, et al. Correlation of cell necrosis and tissue calcification with ischemia/reperfusion injury after liver transplantation. Transplant Proc 2004;36:1766–1768.
Anderson HC . Calcific diseases. A concept. Arch Pathol Lab Med 1983;107:341–348.
Donley GE, Fitzpatrick LA . Noncollagenous matrix proteins controlling mineralization; possible role in pathologic calcification of vascular tissue. Trends Cardiovasc Med 1998;8:199–206.
Davidson D . The function and evolution of Msx genes: pointers and paradoxes. Trends Genet 1995;11:405–411.
Otto F, Lubbert M, Stock M . Upstream and downstream targets of RUNX proteins. J Cell Biochem 2003;89:9–18.
Hanai J, Chen LF, Kanno T, et al. Interaction and functional cooperation of PEBP2/CBF with Smads. Synergistic induction of the immunoglobulin germline Calpha promoter. J Biol Chem 1999;274:31577–31582.
Miyazono K, Maeda S, Imamura T . Coordinate regulation of cell growth and differentiation by TGF-beta superfamily and Runx proteins. Oncogene 2004;23:4232–4237.
Tzimas GN, Afshar M, Chevet E, et al. Graft calcifications and dysfunction following liver transplantation. BMC Surg 2004;4:9.
Miao D, Scutt A . Histochemical localization of alkaline phosphatase activity in decalcified bone and cartilage. J Histochem Cytochem 2002;50:333–340.
Xue Y, Karaplis AC, Hendy GN, et al. Genetic models show that parathyroid hormone and 1,25-dihydroxyvitamin D3 play distinct and synergistic roles in postnatal mineral ion homeostasis and skeletal development. Hum Mol Genet 2005;14:1515–1528.
van der Merwe SW, van den Bogaerde JB, Goosen C, et al. Hepatic osteodystrophy in rats results mainly from portasystemic shunting. Gut 2003;52:580–585.
Miao D, Bai X, Panda D, et al. Osteomalacia in hyp mice is associated with abnormal phex expression and with altered bone matrix protein expression and deposition. Endocrinology 2001;142:926–939.
Miraux S, Lemiere S, Pineau R, et al. Inhibition of FGF receptor activity in glioma implanted into the mouse brain using the tetracyclin-regulated expression system. Angiogenesis 2004;7:105–113.
Nguyen DT, Kebache S, Fazel A, et al. Nck-dependent activation of extracellular signal-regulated kinase-1 and regulation of cell survival during endoplasmic reticulum stress. Mol Biol Cell 2004;15:4248–4260.
Lu Z, Dono K, Gotoh K, et al. Participation of autophagy in the degeneration process of rat hepatocytes after transplantation following prolonged cold preservation. Arch Histol Cytol 2005;68:71–80.
Syntichaki P, Tavernarakis N . Death by necrosis. Uncontrollable catastrophe, or is there order behind the chaos? EMBO Rep 2002;3:604–609.
Watson KE . Pathophysiology of coronary calcification. J Cardiovasc Risk 2000;7:93–97.
Ramadori G, Saile B . Mesenchymal cells in the liver—one cell type or two? Liver 2002;22:283–294.
Corpechot C, Barbu V, Wendum D, et al. Hypoxia-induced VEGF and collagen I expressions are associated with angiogenesis and fibrogenesis in experimental cirrhosis. Hepatology 2002;35:1010–1021.
Shao JS, Cheng SL, Pingsterhaus JM, et al. Msx2 promotes cardiovascular calcification by activating paracrine Wnt signals. J Clin Invest 2005;115:1210–1220.
Rozga J . Hepatocyte proliferation in health and in liver failure. Med Sci Monit 2002;8:RA32–RA38.
Rosario M, Birchmeier W . How to make tubes: signaling by the Met receptor tyrosine kinase. Trends Cell Biol 2003;13:328–335.
Giachelli CM . Vascular calcification mechanisms. J Am Soc Nephrol 2004;15:2959–2964.
Speer MY, Chien YC, Quan M, et al. Smooth muscle cells deficient in osteopontin have enhanced susceptibility to calcification in vitro. Cardiovasc Res 2005;66:324–333.
Wan M, Cao X . BMP signaling in skeletal development. Biochem Biophys Res Commun 2005;328:651–657.
Palcy S, Goltzman D . Protein kinase signalling pathways involved in the up-regulation of the rat alpha1(I) collagen gene by transforming growth factor beta1 and bone morphogenetic protein 2 in osteoblastic cells. Biochem J 1999;343 (Part 1):21–27.
Acknowledgements
We thank Dr Kenneth Finnson, Division of Plastic Surgery, McGill University, Montreal, Canada, for providing anti-TGFβ-1 antibody. This work was supported by grants from the Canadian Institutes for Health Research (CIHR) to EC and DG, National Science and Engineering Research Council (NSERC) to HV. PA was supported by a sabbatical fellowship from Université Bordeaux 1 (Talence, France) and by a grant from the Fondation pour la Recherche Médicale (FRM). EC is a junior scholar from the Fonds de la Recherche en Santé du Québec (FRSQ).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Kalantari, F., Miao, D., Emadali, A. et al. Cellular and molecular mechanisms of abnormal calcification following ischemia–reperfusion injury in human liver transplantation. Mod Pathol 20, 357–366 (2007). https://doi.org/10.1038/modpathol.3800747
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800747
Keywords
This article is cited by
-
Early graft calcification without graft dysfunction after living donor liver transplantation: two case reports
Clinical Journal of Gastroenterology (2021)
-
Calcified desmoplastic fibroblastoma of the foot: imaging findings
Skeletal Radiology (2017)
-
Microarray Profiling of Diaphyseal Bone of Rats Suffering from Hypervitaminosis A
Calcified Tissue International (2012)
