
Abstract
Tumor development from an early lesion through to invasive disease is not a clearly defined progression in the breast. Studies of invasive lobular carcinoma have reported mutations, loss of heterozygosity (LOH) and loss of protein expression in epithelial (E)-cadherin, a protein involved in cell adhesion. Our study examines in situ lobular neoplastic lesions without concurrent invasive carcinoma for E-cadherin gene alterations and protein expression, beta-catenin, alpha-catenin and p120-catenin protein expression, and LOH at the chromosome 16q locus, with the goal of determining the events occurring at the stage of lobular neoplasia. In all, 13 atypical lobular hyperplasia lesions and 13 lobular carcinoma in situ lesions from archived cases were examined. E-cadherin sequence alterations were evaluated using single strand conformation polymorphism and DNA sequencing, and PCR-based LOH analysis was carried out for the 16q locus. Using immunohistochemistry, we assessed protein expression. A total of 23 of 24 lesions evaluated by immunohistochemistry were negative for both E-cadherin and beta-catenin protein expression, and 21 of 23 lesions were negative for alpha-catenin. Cytoplasmic (rather than membrane) localization of p120-catenin was observed in 20 of 21 cases. Lobular carcinoma in situ cases were characterized by mutations; however, atypical lobular hyperplasia cases were not. LOH at 16q was an infrequent event. From our study, we conclude that an altered E-cadherin adhesion complex is an early event affecting atypical lobular hyperplasia as well as lobular carcinoma in situ and occurs prior to progression to invasive disease. However, the loss of protein expression is accompanied by E-cadherin DNA alterations in lobular carcinoma in situ but not in atypical lobular hyperplasia. These cases lacking both protein expression and gene alterations suggest that another mechanism is involved, possibly as early as at the hyperplastic stage, causing silencing of the E-cadherin complex.
Similar content being viewed by others


Main
Tumor development from an early lesion through to invasive disease is not a clearly defined progression in the breast. For invasive breast carcinomas of the lobular histological subtype there is increasing evidence that in situ lobular neoplastic lesions are not only indicators of increased risk but may also act as precursors in the progression to invasive carcinoma.1 Our study investigates in situ lobular neoplastic lesions that have not progressed to invasive disease with the goal of determining the molecular genetic events occurring at the stage of lobular neoplasia. Discovering the specific events that mark the transition from an early lobular neoplastic lesion to an invasive tumor is necessary to both support and subsequently understand this breast cancer progression.
Lobular neoplasia is a histological classification that includes atypical lobular hyperplasia and lobular carcinoma in situ. Lobular carcinoma in situ is a lesion of epithelial origin and is defined by a population of cells that are small, round, monomorphic and discohesive. The lesion is often multicentric and bilateral, and greater than 50% of the acini in the affected terminal duct lobular unit are distended by the cellular proliferation. The cells that define atypical lobular hyperplasia are similar to those that characterize lobular carcinoma in situ, however, the cellular proliferation does not completely occlude the lumen, and less than 50% of the acini in the affected terminal duct lobular unit exhibit distension.2 Both lobular neoplastic lesions are found incidentally during breast tissue biopsy due to their inability to be detected by palpation or mammography.
Histologically, there is a proliferative gradation from lobular hyperplasia to in situ carcinoma, which is also reflected in the relative risk to the patient in the development of invasive disease. A number of epidemiological studies have reported that lobular neoplastic lesions are high-risk indicators.3, 4, 5 A finding of atypical lobular hyperplasia has been reported to imply a four- to five-fold increased risk of subsequent carcinoma in either breast, and a finding of lobular carcinoma in situ implies an eight- to ten-fold increased risk to the patient.
In sporadic breast cancers, histological type has been correlated with expression of the cell adhesion protein epithelial (E)-cadherin, the cadherin subtype expressed in epithelial cells. Proteins that complex with E-cadherin at the cell membrane include beta-, gamma-, alpha- and p120-catenin. Reduced expression of E-cadherin has been reported in invasive ductal carcinoma whereas lobular carcinoma in situ and invasive lobular carcinoma show complete loss of the protein.6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16 A report on invasive lobular carcinomas with adjacent lobular carcinoma in situ demonstrated not only loss of E-cadherin expression but also the simultaneous loss of beta-, gamma- and alpha-catenin protein expression.15 More recently, Sarrio et al17 demonstrated that the loss of E-cadherin along with the cytoplasmic localization of p120-catenin characterizes lobular breast cancers and suggested that p120-catenin plays a role in mediating the oncogenic effects of E-cadherin loss in these cancers. Unquestionably there is evidence that an altered E-cadherin adhesion complex is characteristic of invasive lobular carcinoma and lobular carcinoma in situ with adjacent invasive lobular carcinoma.
In light of these findings, several studies have investigated the E-cadherin gene, CDH1, for alterations in lobular carcinomas. Mutations have been detected in invasive lobular carcinoma and lobular carcinoma in situ with adjacent invasive,6, 11, 12, 13, 14, 15, 16 and the loss of chromosome 16q was detected in solitary in situ and synchronous in situ/invasive lesions.1, 18 These investigations of lobular carcinoma and E-cadherin have provided some evidence that in situ lobular carcinoma may be not only a risk indicator but also a precursor lesion to invasive carcinoma.
To date, most molecular genetic studies of lobular carcinoma in situ have focused on lesions with adjacent invasive carcinoma. However, to study events occurring specifically at the hyperplastic and in situ stages, it is necessary to examine cases where the neoplastic lesion is not contaminated by an invasive lesion. Cases containing atypical lobular hyperplasia and lobular carcinoma in situ without adjacent invasive carcinomas are known to occur in only 0.5–3.8% of breast cases that are otherwise benign.5, 19 These lesions have rarely been studied at the molecular level. We have accrued a collection of cases fitting these criteria, making it possible to determine protein expression and gene alterations occurring at the stage of lobular neoplasia.
Materials and methods
Tissue Accrual
The study population consisted of 21 formalin-fixed, paraffin-embedded, archived cases accrued through the Mount Sinai Hospital Pathology Department (Toronto, ON, Canada). The cases were accessioned from 1988 to 2003. The study pathologist (FOM) reviewed each neoplastic lesion from hematoxylin and eosin (H&E)-stained sections. Previously described histological characteristics20 were used to classify the lobular lesions. The collection included 13 atypical lobular hyperplasia lesions (A1–A13) and 13 lobular carcinoma in situ lesions (L1–L12) lacking adjacent invasive carcinoma. Four of these cases contained both hyperplasia and in situ lesions (A2/L1, A3/L3, A4/L5, A11/L12). More specifically, case A2/L1 was also characterized by multifocal lobular carcinoma in situ (L1-1, L1-2) and these in situ lesions were housed in separate blocks.
Two cases of interest, supplementary to our collection, were also included in all analyses. Case A14/L13 contained atypical lobular hyperplasia and lobular carcinoma in situ lesions as well as a focus of invasive lobular carcinoma in a separate block; the lobular neoplastic lesions were analyzed. Moreover, case P1 contained a lesion of low-grade ductal carcinoma in situ that was used as a control for the immunohistochemistry analyses. All cases were coded to prevent bias and maintain confidentiality.
To determine the robustness of the histological classification of the lesions, all cases were reviewed independently by a second pathologist (ALB) blinded to the results of the molecular analyses. There were discrepancies in two cases. Both cases had been called lobular carcinoma in situ by the first pathologist (FOM) and ‘atypical lobular hyperplasia with duct involvement by cells of atypical lobular hyperplasia’ by the second pathologist (ALB). A consensus diagnosis of lobular carcinoma in situ was reached for both cases by the pathologists following rereview at a multiheaded microscope. Ultimately, the pathology review confirmed that our collection contained 13 atypical lobular hyperplasia and 13 lobular carcinoma in situ lesions without adjacent invasive carcinoma.
Microdissection, DNA Extraction and DNA Amplification
From each case, serial sections (8 μm) were cut from the block containing the lesion of interest and mounted on glass slides. Following deparaffinization, the lesion was removed from the section using either a stereomicroscope-based microdissection technique21 or laser-capture microdissection (PixCell II, Arcturus, CA, USA). The microdissection technique used was determined by the size of the lesion. The majority of the in situ lesions (L1-1, L1-2, L2, L4, L6–L11) were microdissected using the stereomicroscope-based technique due to large lesion area. However, all hyperplasia lesions (A1–A14) were small in size and required laser-capture microdissection in order to accurately remove the lesion from the surrounding tissue. Cases L3, L5, L12 and L13 were also microdissected by laser-capture microdissection because they contained both neoplastic lesions in close proximity in the same section. Figure 1 depicts a case containing atypical lobular hyperplasia from our collection and demonstrates the lesion before and after laser-capture microdissection, as well as the degree of cellularity of a typical atypical lobular hyperplasia lesion for our collection. Whether by stereomicroscope or laser-capture, the use of a microdissection technique allowed for the isolation of a population of lobular neoplastic cells from each atypical lobular hyperplasia or lobular carcinoma in situ lesion that was assured to contain no greater than 15–20% contamination of non-neoplastic cells. Following microdissection, the tissue was incubated for 48 h and DNA was extracted using the QiaAMP DNA Mini Kit (Qiagen, Canada).

An atypical lobular hyperplasia lesion from our collection before and after laser-capture microdissection. (a) A section containing atypical lobular hyperplasia, stained with hematoxylin, before microdissection by the laser-capture microdissection technique. (b) The tissue remaining on the slide following microdissection of the lesion. Using laser-capture microdissection we could maintain the purity of the lesions, with only 15–20% contamination by non-neoplastic cells (intermediate power, × 20).
As neoplastic lesions are small in size, the amount of DNA available per case was limited. To increase the quantity of DNA available for mutation detection, we used the whole genome amplification technique degenerate oligonucleotide-primed polymerase chain reaction (DOP PCR)22 Random degenerate oligonucleotide primers and a modified PCR cycle were used to amplify the added DNA template. Each DOP PCR reaction contained 2 μl of microdissected DNA template and was amplified as per the manufacturer's instructions (DOP PCR Kit, Roche Biomolecular, Canada).
Sequence Alteration Detection and Characterization
To detect sequence alterations, we used single strand conformation polymorphism (SSCP) followed by manual DNA sequencing to characterize each alteration. For the initial screening, each exon of CDH1 was individually amplified using PCR. Exon-specific PCR conditions were optimized for all primer pairs (exons 1–3, 6–166; exons 4–511). Amplification was performed in a volume of 30 μl containing 10 μl of DOP PCR product template, 1 × High Fidelity PCR Buffer, 2 mM MgSO4, 0.2 mM of each dNTP, 0.3 μM of forward and reverse primers, 1 U of PLATINUM Taq DNA polymerase High Fidelity (GIBCO BRL, Life Technologies, Canada). and 0.1 μCi of 33P(Phosphorus-33)-labeled dATP (Perkin-Elmer, USA). Following an initial denaturation step at 94°C for 3 min, 40 cycles of 94°C for 15 s, 50°C (exons 2, 3, 7–10, 16), or 53°C (exon 6), or 55°C (exons 4 and 5), or 58°C (exons 11–15), or 68°C (exon 1) for 15 s, and 72°C for 20 s were performed.
A stop solution (95% formamide, 20 mM EDTA, 0.05% bromophenol blue, 0.05% xylene cyanol FF) was added to the SSCP reactions, which were subsequently heat denatured and subjected to electrophoresis on an 8% nondenaturing polyacrylamide gel (including 10% glycerol). Two gels were run simultaneously under different conditions: (i) 4°C, 8 W for 12 h, and (ii) room temperature, 12 W for 16 h. Results were obtained following autoradiography. If an aberrantly migrating band was observed, the SSCP reaction was repeated for that case using 5 μl of microdissected DNA template that had not been subjected to DOP PCR amplification (PCR/SSCP conditions as previously stated). If the abnormal banding pattern could be duplicated in this second independent SSCP experiment, then the alteration was characterized using manual DNA sequencing.
To characterize CDH1 alterations, aberrantly migrating bands were excised from the dried SSCP gel and DNA was extracted using a serial freeze–thaw technique. In all, 5 μl of the extracted DNA was used as template in an exon-specific PCR reaction using the conditions previously outlined. The Thermo Sequenase Radiolabelled Terminator Cycle Sequencing Kit (Amersham Pharmacia Biotech, Canada) was used to manually sequence the DNA, according to the manufacturer's instructions. Results were obtained following autoradiography. Alterations that were found were confirmed by repeating the sequencing using microdissected DNA (not previously preamplified by DOP PCR) as template from both the forward and reverse direction for each exon.
Loss of Heterozygosity (LOH)
LOH was evaluated for the E-cadherin gene located on chromosome 16q. Five microsatellite markers, located at chromosome locus 16q21–16q22.1, were used (D16S421, D16S496, D16S503, D16S3095, D16S752). Microdissected DNA from each lesion (not preamplified by DOP PCR), paired with DNA from an adjacent area of normal tissue, was used as template to examine LOH.
PCR amplification was performed in a volume of 30 μl containing 5 μl of microdissected DNA template, 1 × High Fidelity PCR Buffer, 2 mM MgSO4, 0.2 mM of each dNTP, 0.3 μM of forward and reverse primers, 1 U of PLATINUM Taq DNA polymerase High Fidelity (GIBCO BRL, Life Technologies, Canada). and 0.1 μCi of [33P]dATP (Perkin-Elmer, USA). Following an initial denaturation step at 94°C for 3 min, 40 cycles of 94°C for 15 s, 54°C (D16S503) or 57°C (D16S421, D16S496, D16S752, D16S3095) for 15 s, and 72°C for 20 s were performed. A stop solution (as previously described) was added and each reaction was subsequently heat denatured and subjected to electrophoresis on a 7% denaturing formamide gel, which was run at 80 W for 3 h. Results were obtained following autoradiography. Multiple independent observers evaluated each marker, scoring each case as ‘LOH’, ‘no LOH’, or ‘uninformative’. To evaluate each marker, LOH was defined as a relative decrease in band intensity greater than 50%. For each case, a minimum of three of the five markers with observed LOH was required for an overall classification of LOH.
Immunohistochemistry
Following sectioning for microdissection, 4 μm sections were cut for each formalin-fixed, paraffin-embedded block, and mounted on glass slides. Each section was deparaffinized in xylene and rehydrated through graded alcohols to distilled water. Following heat antigen retrieval, the primary antibodies to E-cadherin (HECD-1, Monoclonal Mouse anti-E-cadherin 2nd Gen Predilute Antibody, Zymed Laboratories Inc., USA), beta-catenin (Monoclonal Mouse anti-beta-catenin, 1:6000 dilution, Transduction Laboratories, USA), alpha-catenin (NCL-A-CAT, Monoclonal Mouse anti-alpha-catenin, 1:50 dilution, Novocastra Laboratories Ltd, UK) or p120-catenin (p120-ctn (15D2), 1:50 dilution, Santa Cruz Biotechnology, Inc., USA) were applied. The Ultra Streptavidin Detection System (Signet Laboratories Inc., USA) was used as per the manufacturer's instructions for all antibodies except alpha-catenin, which required the ELITE Detection System (Vector Laboratories (Canada) Inc., Canada). Each section was developed with the chromogen diaminobenzidene and sections were counterstained in hematoxylin.
Some cases have insufficient material to carry out immunohistochemistry due to sectioning order. Sections were cut from each case for immunohistochemistry analyses only after sectioning was complete for microdissection. In some cases, the lesion of interest was no longer present in the immunohistochemistry section and therefore no result could be obtained.
Immunohistochemical staining was reviewed and scored by the study pathologist. To evaluate the immunohistochemistry for E-cadherin, beta-catenin and alpha-catenin protein expression, a positive stain was determined to be complete circumferential membrane staining of the lobular neoplastic cells. The case of low-grade ductal carcinoma in situ (P1) was used as a positive control for E-cadherin, beta-catenin and alpha-catenin staining as it expressed these proteins at the membrane. Evaluation of the immunohistochemistry for p120-catenin required assessment of the circumferential membrane staining as well as the cytoplasmic staining pattern. A formalin-fixed, paraffin-embedded breast cancer cell line (MCF7) was used as the positive control for the p120-catenin immunohistochemistry as it contained membrane localized p120-catenin.
Results
CDH1 Mutation Analysis
Using the manual DNA sequencing technique, 16 polymorphisms (data not shown) and 15 mutations (Table 1) were characterized. Three mutations (cases A12, L4, L8) were deletions causing a frameshift and a premature stop codon. In all, 11 sequence alterations were classified as missense mutations (cases L1–1, L1–2, L2, L3, L5, L6, L7, L9, L10, L11, L12). The mutations were found in exons 3, 6, 7, 9, 10, 11, 12, 13 and 15. With the exception of the missense mutation found in case L1–1 (previously reported by Rieger-Christ et al11), all alterations found in this study are novel.
Four cases (A2/L1, A3/L3, A4/L5, A11/L12) contained both atypical lobular hyperplasia and lobular carcinoma in situ lesions. Each of these cases of lobular carcinoma in situ contained a sequence alteration that was not detected in the adjacent hyperplastic lesions. In addition to containing both types of lesions, case A2/L1 contained, in separate blocks, two lobular carcinoma in situ lesions (L1–1, L1–2). These in situ lesions were microdissected individually and each was found to harbor different missense mutations.
In order to predict if the missense mutations found in our cases of lobular carcinoma in situ would have a phenotypic effect we used SIFT (http://blocks.fhcrc.org/sift/SIFT.html), a sequence homology-based tool. Of the 11 missense mutations, three were predicted by SIFT not to be tolerated amino-acid substitutions (case L5, L9, L10).
Case A14/L13, noted as containing adjacent atypical lobular hyperplasia and lobular carcinoma in situ lesions as well as invasive lobular carcinoma in a separate block, was found to contain a nonsense mutation in exon 11. As observed in the four cases containing both lobular neoplastic lesions, the sequence alteration is present in the in situ component but not in the hyperplasia. Figure 2 shows the mutation found in L13 and the corresponding sequence from the adjacent hyperplasia (A14) lacking the alteration.

Characterization of CDH1 sequence alterations and LOH at 16q. (a) Sequence alteration found in case A14/L13. The alteration was found in the lobular carcinoma in situ lesion (sequence on the right) and not the atypical lobular hyperplasia lesion (sequence on the left). The autoradiograph shows the missense mutation (1595G>A) that translates into an amino-acid change of Trp to amber, which causes a stop in the sequence in exon 11 of CDH1. (b) Case L7 (denoted as L-1, N-1) contains LOH and case A11/L12 (denoted as A-2, L-2, N-2) does not. LOH was determined using DNA from the lesions paired with adjacent normal tissue. The lesions are labeled as ‘A’ for atypical lobular hyperplasia, ‘L’ for lobular carcinoma in situ, and ‘N’ for corresponding normal epithelium.
Loss of Heterozygosity
LOH was evaluated with five microsatellite markers and each case was evaluated paired with its corresponding normal for each marker (Table 1). We observed 80–100% agreement between observers and questionable cases were repeated and reevaluated. Three cases had insufficient material to carry out LOH analysis (A3, A6, L9). Of the remaining cases, three were found to have LOH (A10, L1–2, L7) and 20 showed no LOH (Figure 2).
E-cadherin, Beta-catenin, Alpha-catenin and p120-catenin Protein Expression
All lobular carcinoma in situ and 11 of 12 atypical lobular hyperplasia lesions were negative for E-cadherin and beta-catenin staining (Table 1, Figure 3); case A7 showed positive staining of the lobular neoplastic cells despite its identical morphologic appearance to the other cases of atypical lobular hyperplasia. The lesions in case A14/L13 had an identical staining pattern to all the lobular neoplastic cases, with negative E-cadherin and beta-catenin protein expression. For alpha-catenin, 10 of 11 lobular carcinoma in situ and 11 of 12 atypical lobular hyperplasia lesions were scored as negative, and two cases (A7, L8) were positive. The case of low-grade ductal carcinoma in situ (P1) stained positive for E-cadherin, beta-catenin and alpha-catenin. Moreover, all cases contained adjacent normal epithelium that served as an internal positive control and in all cases showed complete circumferential membrane staining (Figure 4).

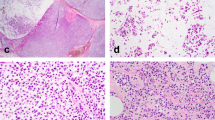
Examination of E-cadherin, beta-catenin, alpha-catenin and p120-catenin protein expression by immunohistochemistry. (a) Case A2, containing atypical lobular hyperplasia, stained with H&E to show cellular architecture. The corresponding negative membrane staining for (b) E-cadherin (some background spotty cytoplasmic staining is observed with this antibody), (c) beta-catenin, (d) alpha-catenin and (e) p120-catenin stained sections from this case. The p120-catenin stain shows cytoplasmic localization of the protein. (f) An H&E-stained section from a case containing lobular carcinoma in situ (case L2); and the corresponding negative membrane staining for (g) E-cadherin, (h) beta-catenin, (i) alpha-catenin and (j) p120-catenin for this case. The p120-catenin staining of the in situ case shows cytoplasmic localization with some accentuation in the perinuclear zone. (k) An H&E-stained lesion of low-grade ductal carcinoma in situ (case P1), used as a positive control for the (l) E-cadherin, (m) beta-catenin, and (n) alpha-catenin immunohistochemistry experiments. (o) Formalin-fixed, paraffin-embedded MCF7 breast cancer cell line stained for p120-catenin and used as a positive control for p120-catenin immunohistochemistry (high power, × 40).

Normal breast acini stained by immunohistochemistry. All cases of atypical lobular hyperplasia and lobular carcinoma in situ contained areas of normal nonproliferating acini, adjacent to the lesion of interest, which served as internal positive control (Intermediate power, × 20). (a) Case L2 stained for E-cadherin protein expression, with complete circumferential membranous staining in the area of normal epithelium. (b) Case L2 stained for beta-catenin, showing adjacent normal acini with complete circumferential membranous staining. (c) Case A11/L12 stained for alpha-catenin, with adjacent normal acini showing complete circumferential membranous staining. (d) Case A5 stained for p120-catenin demonstrates cytoplasmic localization of the protein in the lesion and complete circumferential membranous staining in the adjacent normal acini (inset: high power, × 40).
The formalin-fixed, paraffin-embedded breast cancer cell line (MCF7) showed membrane localization of p120-catenin, and therefore positive circumferential membrane staining. Conversely, all lobular carcinoma in situ lesions showed no membrane, but diffuse cytoplasmic staining, as did 10 of 11 atypical lobular hyperplasia lesions, and only one case (A7) showed complete circumferential membrane staining.
Discussion
We investigated atypical lobular hyperplasia and lobular carcinoma in situ lesions for E-cadherin gene alterations and protein expression, beta-, alpha-, and p120-catenin protein expression, and LOH at the chromosome 16q locus. Unlike most studies, our atypical lobular hyperplasia and lobular carcinoma in situ cases were specifically selected without adjacent invasive carcinoma. The information to be gained from studying these lobular neoplastic lesions is substantial when considering the ambiguous understanding of the molecular genetic events occurring at these early stages.
A number of studies have shown that E-cadherin is completely inactivated in invasive lobular carcinoma. Definitively, from our study of neoplastic lesions, we can conclude that lobular lesions, whether hyperplasia or carcinoma in situ, lack E-cadherin membrane staining. These immunohistochemistry results support the previously reported correlation between protein inactivation and histological type. Furthermore, the data indicate that this correlation is not restricted to lobular carcinoma in situ and invasive lobular carcinoma but can be extended to atypical lobular hyperplasia as well.
In addition, a complete lack of beta-catenin and alpha-catenin protein expression as well as cytoplasmic localization of p120-catenin was characteristic to the lobular neoplastic cells in all but one case. We postulate that this one exceptional case (A7), with membrane localization of all proteins of the E-cadherin complex, has not yet undergone the molecular genetic events that cause inactivation of the E-cadherin complex. Irrespective of case A7, the results of our immunohistochemical analyses demonstrate that without the presence of an invasive lesion the expression of the entire E-cadherin complex at the cell membrane is altered in both atypical lobular hyperplasia and lobular carcinoma in situ lesions.
The use of a whole genome amplification technique to increase the quantity of the DNA template obtained from the lobular neoplastic lesions made it possible to complete the screening of CDH1 for sequence alterations. DOP PCR has been used previously in combination with SSCP and is sensitive with respect to amplifying small quantities of DNA.23 According to us CDH1 mutation analysis by DOP PCR-SSCP was reliable, as aberrantly migrating bands found by using this technique could be duplicated by SSCP using microdissected DNA, without prior amplification by DOP PCR, as template.
Only one (A12) of 13 atypical lobular hyperplasia lesions contained an alteration. On the other hand, every case of lobular carcinoma in situ analyzed has been found to harbor a sequence alteration (13/13). To date, there have been no mutations reported in cases of lobular carcinoma in situ that lacked adjacent invasive carcinoma; somatic mutations have only been found in invasive lobular carcinoma or lobular carcinoma in situ with adjacent invasive disease. Although the data do not allow us to speculate as to whether these in situ lesions are precursors to invasive carcinoma, we can conclude that somatic alterations in CDH1 appear to occur predominantly at the in situ stage.
The study design, the sensitivity of the techniques used and the high cellularity of the lesions of atypical lobular hyperplasia ruled out the possibility of false-negative results with respect to CDH1 mutation analysis. The microdissection techniques allowed for the isolation of each lesion from the surrounding tissue/adjacent lesions with no greater than 15–20% contamination of non-neoplastic cells (Figure 1). In addition, the lack of sequence alterations found in the hyperplasia cases was reproduced in independent experiments using microdissected DNA (not previously preamplified by DOP PCR) from each atypical lobular hyperplasia lesion. Altogether the trend that lobular carcinoma in situ but not atypical lobular hyperplasia cases carry alterations was prominent. Furthermore, this trend supports previous reports of a precursor role for in situ lesions as it demonstrates an increase in genetic hits from hyperplasia to carcinoma in situ characteristic of progression.
Moreover, in the cases that contained both lobular neoplastic lesions, the atypical lobular hyperplasia and lobular carcinoma in situ were microdissected separately and in all cases the in situ component was found to harbor a sequence alteration, whereas hyperplasia did not. Even case A14/L13, noted in the pathology review as containing adjacent atypical lobular hyperplasia and lobular carcinoma in situ lesions as well as invasive lobular carcinoma in a separate block, was found to contain a mutation in the in situ stage but not hyperplasia. These cases further substantiate our hypothesis that mutations are first detected at the in situ stage.
CDH1 sequence alterations reported to date in studies investigating progression from lobular carcinoma in situ to invasive lobular carcinoma have been inactivating mutations. The nonsense mutation found in case A14/L13 was predicted to cause protein truncation. As this case (A14/L13) has an adjacent invasive lesion, we suggest that the presence of an inactivating CDH1 mutation could be an event that distinguishes lobular carcinoma in situ lesions that are precursors from those that are not.
The frameshift mutations found in cases L4, L8 and A12 are likely to have an effect on protein function. The bioinformatics tool, SIFT, clarified to some extent the functional significance of the missense mutations we detected. As only three missense mutations were predicted to affect protein function, it is likely that lobular carcinoma in situ lesions found to harbor only missense mutations do not progress to invasive carcinoma. The presence of a missense mutation could simply indicate an environment amenable to genetic alteration, as observed in case A2/L1 where adjacent in situ lesions were found to harbor different missense mutations. Moreover, since the loss of protein expression was not always associated with a sequence alteration, as in the cases containing atypical lobular hyperplasia, we conclude that mutation alone could not cause the lack of E-cadherin protein expression that we have observed.
Very little is known about the molecular genetic events occurring at the stage of atypical lobular hyperplasia. To our knowledge, this study represents the first investigation of alterations in E-cadherin in atypical lobular hyperplasia. The overwhelming absence of mutations in cases of atypical lobular hyperplasia, coupled with a loss of E-cadherin protein expression, suggests that in these lesions, E-cadherin may be inactivated by means other than the presence of mutation. To address this, we evaluated these lesions for LOH.
LOH has been studied in lobular breast cancers and the chromosomal region of 16q, the location of CDH1,24 has been found to have a high degree of LOH.6, 12, 13, 16 These previous studies have found LOH to accompany mutations in cases of invasive lobular carcinoma or lobular carcinoma in situ with adjacent invasive lobular carcinoma. However, in the present study, all cases of lobular carcinoma in situ were found to harbor mutations but LOH was found in only two of these 13 cases. In both instances, LOH was detected in cases that harbored missense mutations. The classic pattern of an inactivating mutation coupled with LOH does not appear to be characteristic of the lobular carcinoma in situ lesions in our collection.
Methylation of the E-cadherin promoter has been reported in studies investigating invasive lobular carcinoma,16 breast carcinomas,25 and breast cancer cell lines that lack E-cadherin expression.26, 27 In the case of sporadic gastric carcinomas, promoter methylation has been described as a second hit leading to inactivation of the E-cadherin gene.28 We hypothesize that epigenetic mechanisms acting at the hyperplastic stage could provide an explanation for the loss of E-cadherin that we have observed in both atypical lobular hyperplasia and lobular carcinoma in situ. Further research efforts will address this possibility.
Although many epidemiological studies have clarified the risk associated with atypical lobular hyperplasia and lobular carcinoma in situ, at present there are no morphological or clinical features that help identify those individuals who have the greatest risk of developing invasive disease. From our study, we conclude that an altered E-cadherin adhesion complex, including alpha-, beta- and p120-catenin, is characteristic of lobular neoplastic lesions and occurs prior to progression to invasive disease. Furthermore, somatic mutations appear to be an event characteristic of lobular carcinoma in situ and not atypical lobular hyperplasia lesions, and we suggest inactivating mutations could possibly distinguish the lobular carcinoma in situ lesions that may progress to invasive disease. However, the reported molecular data, that is, mutations, LOH, chromosomal gains/losses, and loss of protein expression, coupled with the findings of our investigation, still leave questions regarding the progression from lobular neoplasia to invasive breast cancer. Further research into the molecular events occurring at the hyperplastic and in situ stages is essential to understanding and identifying this subset of lobular neoplastic lesions that have the highest risk of progressing to invasive carcinoma.
References
Shelley Hwang E, Nyante SJ, Yi Chen Y, et al. Clonality of lobular carcinoma in situ and synchronous invasive lobular carcinoma. Cancer 2004;100:2562–2572.
Lishman SC, Lakhani SR . Atypical lobular hyperplasia and lobular carcinoma in situ: surgical and molecular pathology. Histopathology 1999;35:195–200.
Page DL, Schuyler PA, Dupont WD, et al. Atypical lobular hyperplasia as a unilateral predictor of breast cancer risk: a retrospective cohort study. Lancet 2003;361:125–129.
London SJ, Connolly JL, Schnitt SJ, et al. A prospective study of benign breast disease and the risk of breast cancer. JAMA 1992;267:941–944.
Page DL, Kidd Jr TE, Dupont WD, et al. Lobular neoplasia of the breast: higher risk for subsequent invasive cancer predicted by more extensive disease. Hum Pathol 1991;22:1232–1239.
Berx G, Cleton-Jansen AM, Strumane K, et al. E-cadherin is inactivated in a majority of invasive human lobular breast cancers by truncation mutations throughout its extracellular domain. Oncogene 1996; 13:1919–1925.
Gamallo C, Palacios J, Suarez A, et al. Correlation of E-cadherin expression with differentiation grade and histological type in breast carcinoma. Am J Pathol 1993;142:987–993.
Moll R, Mitze M, Frixen UH, et al. Differential loss of E-cadherin expression in infiltrating ductal and lobular breast carcinomas. Am J Pathol 1993;143:1731–1742.
Goldstein NS, Bassi D, Watts JC, et al. E-cadherin reactivity of 95 noninvasive ductal and lobular lesions of the breast. Implications for the interpretation of problematic lesions. Am J Clin Pathol 2001;115:534–542.
Goldstein NS, Kestin LL, Vicini FA . Clinicopathologic implications of E-cadherin reactivity in patients with lobular carcinoma in situ of the breast. Cancer 2001;92:738–747.
Rieger-Christ KM, Pezza JA, Dugan JM, et al. Disparate E-cadherin mutations in LCIS and associated invasive breast carcinomas. Mol Pathol 2001;54:91–97.
Berx G, Cleton-Jansen AM, Nollet F, et al. E-cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. EMBO J 1995;14:6107–6115.
Huiping C, Sigurgeirsdottir JR, Jonasson JG, et al. Chromosome alterations and E-cadherin gene mutations in human lobular breast cancer. Br J Cancer 1999;81:1103–1110.
Vos CB, Cleton-Jansen AM, Berx G, et al. E-cadherin inactivation in lobular carcinoma in situ of the breast: an early event in tumorigenesis. Br J Cancer 1997;76:1131–1133.
De Leeuw WJ, Berx G, Vos CB, et al. Simultaneous loss of E-cadherin and catenins in invasive lobular breast cancer and lobular carcinoma in situ. J Pathol 1997;183:404–411.
Droufakou S, Deshmane V, Roylance R, et al. Multiple ways of silencing E-cadherin gene expression in lobular carcinoma of the breast. Int J Cancer 2001; 92:404–408.
Sarrio D, Perez-Mies B, Hardisson D, et al. Cytoplasmic localization of p120ctn and E-cadherin loss characterize lobular breast carcinoma from preinvasive to metastatic lesions. Oncogene 2004;23:3272–3283.
Etzell JE, Devries S, Chew K, et al. Loss of chromosome 16q in lobular carcinoma in situ. Hum Pathol 2001;32:292–296.
Haagensen CD, Lane N, Lattes R, et al. Lobular neoplasia (so-called lobular carcinoma in situ) of the breast. Cancer 1978;42:737–769.
Dupont WD, Page DL . Risk factors for breast cancer in women with proliferative breast disease. N Engl J Med 1985;312:146–151.
Done SJ, Arneson NC, Ozcelik H, et al. p53 mutations in mammary ductal carcinoma in situ but not in epithelial hyperplasias. Cancer Res 1998;58:785–789.
Telenius H, Carter NP, Bebb CE, et al. Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics 1992;13:718–725.
Barbaux S, Poirier O, Cambien F . Use of degenerate oligonucleotide primed PCR (DOP-PCR) for the genotyping of low-concentration DNA samples. J Mol Med 2001;79:329–332.
Berx G, Staes K, van Hengel J, et al. Cloning and characterization of the human invasion suppressor gene E- cadherin (CDH1). Genomics 1995;26:281–289.
Nass SJ, Herman JG, Gabrielson E, et al. Aberrant methylation of the estrogen receptor and E-cadherin 5′ CpG islands increases with malignant progression in human breast cancer. Cancer Res 2000;60:4346–4348.
Graff JR, Herman JG, Lapidus RG, et al. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res 1995;55:5195–5199.
Hiraguri S, Godfrey T, Nakamura H, et al. Mechanisms of inactivation of E-cadherin in breast cancer cell lines. Cancer Res 1998;58:1972–1977.
Machado JC, Oliveira C, Carvalho R, et al. E-cadherin gene (CDH1) promoter methylation as the second hit in sporadic diffuse gastric carcinoma. Oncogene 2001;20: 1525–1528.
Acknowledgements
This work was supported by the Canadian Breast Cancer Research Initiative Grant (009177) to IL Andrulis, and the United States Army Medical Research and Material Command predoctoral fellowship (DAMD 17-02-1-0498) to TL Mastracci. We wish to thank Dr Nalan Gokgoz, Sasha Eskandarian and Nona Arneson for helpful discussions and invaluable technical support throughout these studies; the laboratory technicians in the Pathology Department at Mount Sinai Hospital; and Drs Susan J Done and Louise A Quenneville for assistance with case accrual.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mastracci, T., Tjan, S., Bane, A. et al. E-cadherin alterations in atypical lobular hyperplasia and lobular carcinoma in situ of the breast. Mod Pathol 18, 741–751 (2005). https://doi.org/10.1038/modpathol.3800362
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800362
Keywords
This article is cited by
-
Molecular profile of atypical hyperplasia of the breast
Breast Cancer Research and Treatment (2018)
-
Current Management of High-Risk Breast Lesions
Current Radiology Reports (2018)
-
PIK3CA mutations are common in lobular carcinoma in situ, but are not a biomarker of progression
Breast Cancer Research (2017)
-
Invasive lobular carcinoma of the breast: morphology, biomarkers and ’omics
Breast Cancer Research (2015)
