
Abstract
Barrett's esophagus is a complication of chronic gastroesophageal reflux disease and can be diagnosed when there is an endoscopic abnormality in which a biopsy shows evidence of specialized columnar epithelium, characterized by the presence of acid mucin-containing goblet cells. Much of the controversy in this body of literature relates to the complex anatomy of the esophagogastric junction and the difficulty in precisely identifying this landmark at endoscopy. By definition, in Barrett's esophagus, the squamocolumnar junction is proximal to the esophagogastric junction. Although fundic-type or cardiac-type (junctional) columnar epithelium may be present in Barrett's esophagus, it is only the presence of specialized columnar epithelium that is diagnostic of this condition. Patients with Barrett's esophagus are at risk of progressing to esophageal dysplasia and adenocarcinoma. There are several problems with using dysplasia as a marker for increased cancer risk in these patients, including problems with sampling error and intra- and interobserver variation in the recognition of dysplasia. It may be difficult to distinguish regenerative epithelial changes from dysplasia, low-grade from high-grade dysplasia, and high-grade dysplasia from intramucosal adenocarcinoma. Finally, there are relatively few prospective data evaluating the natural history of high-grade dysplasia. The management of patients with Barrett's-related dysplasia is controversial and varies from institution to institution. Future emphasis should be on cost-effective techniques for sampling as much of the esophageal mucosa as possible in patients who are at the highest risk of progressing to dysplasia and adenocarcinoma. Identification of biomarkers that identify such patients before the histologic recognition of dysplasia will be an area of intensive research.
Similar content being viewed by others
Introduction
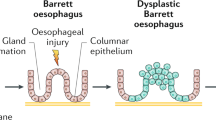
Barrett's esophagus (BE) is a complication of chronic gastroesophageal reflux and results in the replacement of the normal stratified squamous epithelium of the esophagus with columnar epithelium of various types (1). The importance of diagnosing BE is related to its association with the subsequent development of esophageal adenocarcinoma (2), the frequency of which has rapidly increased over the past several decades (3, 4).
In order to properly diagnose BE, it is important to understand the normal anatomy and histology of the esophagogastric junction (EGJ), including the lower esophageal sphincter (LES). The muscular EGJ is the site at which the most distal portion of the esophagus (the distalmost segment of the LES) meets the proximal stomach. Endoscopically, one can closely approximate the muscular EGJ by identifying the proximal margin of the gastric folds (5). The mucosal EGJ, also known as the mucosal squamocolumnar junction (SCJ) or Z-line, is the site at which the squamous mucosa of the esophagus meets columnar-lined mucosa. It is important to understand, however, that the SCJ may be at the same level as the muscular EGJ or may lie 1–2 cm above the muscular EGJ in “normal” individuals. Thus, cardiac and fundic-type mucosae may be found within the distal few centimeters of the esophagus in normal individuals and are presumably related to physiologic reflux. Several studies from the group at the University of Southern California have shown that metaplastic cardiac-type mucosa in the distal esophagus is quite common and is strongly associated with acid reflux (6, 7, 8).
THE NORMAL GASTRIC CARDIA: FACT OR FICTION?
The very existence of the gastric cardia as a normal structure has been the source of great controversy. Most textbooks describe the gastric cardia as a narrow strip of mucosa that separates the most distal portion of the esophageal squamous mucosa from the acid-producing fundic mucosa (9). However, recent data from the University of Southern California group suggest that the gastric cardia is not a normal structure but rather that cardiac-type mucosa is metaplastic (6, 7, 8, 10). Recently, Kilgore et al. (11) evaluated the entire EGJ in 30 pediatric autopsies from patients ≤18 years of age with no known history of GERD or BE. In each case, the SCJ and its relationship to the EGJ was meticulously noted. In all cases, cardiac mucosa was present, always on the gastric side of the EGJ, although it was quite small, ranging from only 1 to 4 mm in length. The results of this study support the concept that the gastric cardia is present from birth as a normal structure, although they do not preclude the possibility that cardiac-type mucosa can arise in the distal esophagus as a metaplastic phenomenon, as proposed by others.
THE ENDOSCOPIC DIAGNOSIS OF BE
Endoscopically, it may be difficult for the gastroenterologist to definitively identify the presence of BE for several reasons (12). First, the presence of a hiatal hernia, a frequent accompaniment of BE, makes identification of the muscular EGJ difficult. Furthermore, there are no anatomic landmarks that clearly define the region of the LES. Thus, it may not be known either to the endoscopist or the pathologist precisely where a biopsy specimen may have come from in relation to the EGJ. A biopsy from the vicinity of the EGJ with intestinal metaplasia could either represent BE or intestinal metaplasia of the most proximal portion of the stomach.
THE HISTOLOGIC DIAGNOSIS OF BE
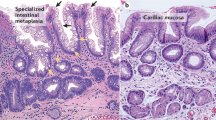
In 1976, Paull et al. (13) described three different types of epithelium in BE: fundic-type, cardiac-type (junctional) and specialized columnar epithelium. The cardiac and fundic types of Barrett's epithelium resemble their normal counterparts in the stomach, except for the presence of some degree of mucosal distortion, glandular atrophy, and mild inflammation (13, 14). A biopsy from the “distal esophagus” with either of these mucosae is not diagnostic of BE because, as stated earlier, these mucosae are frequently found in the distal esophagus in the absence of intestinal metaplasia (9, 10, 11). If the endoscopic impression is clearly that of BE, then the absence of intestinal metaplasia may simply be a function of sampling error. Thus, although the pathologist may not be able to make a definitive diagnosis of BE in this situation, the endoscopic impression may still strongly suggest this diagnosis. Fortunately, this problem is relatively rare, as Weinstein et al. (15) found nonintestinal tongues of columnar epithelium extending >2 cm into the lower esophagus in <1% of 250 cases of BE that were studied. If one performs an Alcian blue stain (pH 2.5), one may find isolated goblet cells that were not readily identifiable on hematoxylin and eosin-stained sections in areas that otherwise resemble cardiac or fundic-type mucosae. When these mucosae are found in a patient who is known to have BE, they are typically present in the most distal portion of the Barrett's segment, often with the following zones: specialized columnar epithelium, found most proximally; fundic-type mucosa, found most distally; and cardiac-type mucosa in between, although a mosaic pattern may also be identified (13).
The presence of specialized columnar epithelium, characterized by acid mucin-containing goblet cells, has been accepted as diagnostic of BE, regardless of the precise site of the biopsy within the tubular esophagus (Figs. 1, 2, 3 and 4; 14, 15). In fact, the American College of Gastroenterology and its Practice Parameters Committee recently provided a definition of BE as “a change in the esophageal epithelium of any length that can be recognized at endoscopy and is confirmed to have intestinal metaplasia by biopsy” (16). BE can be further (arbitrarily) divided into short- (SSBE) and long- (LSBE) segment BE on the basis of the length of esophageal intestinal metaplasia: <3 cm and ≥3 cm, respectively (17, 18).

Barrett's esophagus, characterized by the presence of specialized columnar epithelium with goblet cells.

Alcian blue and periodic acid-Schiff stain of a segment of Barrett's esophagus. The goblet cells are intensely Alcian blue positive because of the presence of acid mucin. The cells between the goblet cells are periodic acid-Schiff positive because of the presence of neutral mucin. This is so-called incomplete intestinal metaplasia.

Complete intestinal metaplasia in a segment of Barrett's esophagus. The epithelium resembles small intestinal epithelium. Goblet cells are readily identified.

Alcian blue and periodic acid-Schiff stain of Barrett's mucosa with complete intestinal metaplasia. The goblet cells are intensely Alcian blue positive. The periodic acid-Schiff portion of the stain outlines a primitive luminal brush border.
Histologically, specialized columnar epithelium is characterized by two cell types: goblet cells and columnar cells. Cytologically, goblet cells have distended, mucin-filled cytoplasm with a barrel-shaped configuration. Histochemically, goblet cells contain acid mucins (both sialo and sulfated mucins), which stain positively with Alcian blue at pH 2.5. The columnar cells in between the goblet cells may resemble either gastric foveolar cells or intestinal absorptive cells, at least at the light microscopic level. Unlike normal gastric foveolar cells, which contain neutral mucin, the columnar cells in BE may contain Alcian blue-positive acid mucin (“the columnar blues”; Fig. 5), although the intensity of staining is not as intense as in the goblet cells (19). Such cells should not be used as definitive evidence of BE because unequivocal goblet cells are required for this diagnosis.

The “columnar blues.” Some of the cells on the surface epithelium show Alcian blue positivity, but none of these cells have the morphologic features of goblet cells. This is not diagnostic of Barrett's esophagus.
BE: WHAT IS THE RISK OF PROGRESSION TO DYSPLASIA AND ADENOCARCINOMA?
Although all patients with BE are at an increased risk for developing adenocarcinoma, certain patients are at higher risk than others. For example, epidemiologic data suggest that the majority of patients with BE-associated adenocarcinoma are older white males (1, 20). There is also evidence to support the contention that only those patients with specialized columnar epithelium are at an increased risk of developing Barrett's-related adenocarcinoma (16, 21, 22, 23). The presence of epithelial dysplasia, particularly high-grade dysplasia (HGD), is also a risk factor for synchronous or metachronous adenocarcinoma (24, 25, 26). Several retrospective studies have noted the frequency with which dysplasia is seen both adjacent to and distant from Barrett's-related adenocarcinomas (1). Prospective studies have also documented the progression from specialized columnar epithelium to HGD and, eventually, invasive adenocarcinoma (23, 27). Thus, dysplasia is not only a marker of adenocarcinoma but clearly is the preinvasive lesion. Finally, although it is known that adenocarcinoma can arise in extremely short segments of BE (28), some workers have proposed that there is an increased risk of adenocarcinoma as the length of the BE increases (29, 30).
CANCER SURVEILLANCE IN BE
Although cancer surveillance is performed in most institutions once a diagnosis of BE is rendered, the true cost-benefit ratio of this endeavor is still essentially unknown. In other words, does the increased risk of adenocarcinoma in these patients justify the cost of a cancer surveillance program, particularly one that has so many inherent problems, as will be discussed below? Although this issue has yet to be resolved, at our institution, patients are placed into a cancer surveillance program once a diagnosis of BE has been clearly established, with the surveillance goal being the identification of epithelial dysplasia in a biopsy specimen, before carcinoma has intervened. We essentially follow the protocol proposed by Reid et al. (24), with four-quadrant biopsies taken at intervals of ≤2 cm throughout the length of the Barrett's segment, with additional biopsies of any endoscopic lesions, using jumbo forceps (31).
HISTOPATHOLOGIC DIAGNOSIS OF DYSPLASIA
Dysplasia can be defined as the presence of neoplastic epithelium that is confined within the basement membrane of the gland within which it arises (32). Unlike inflammatory bowel disease-associated dysplastic lesions, most cases of Barrett's-related dysplasia do not closely resemble colonic adenomas. Rather, the typical form of Barrett's-related dysplasia often arises in glands that retain their normal configuration and often lack nuclear stratification. Using the criteria defined by Riddell et al. (32) for dysplasia arising in inflammatory bowel disease, dysplasia in BE can be classified as either low-grade or high-grade based on the degree of the abnormality present. Thus, the possibilities are as follows: (1) negative for dysplasia; (2) positive for dysplasia, either low-grade or high-grade; or (3) indefinite for dysplasia.
In low-grade dysplasia (LGD), crypt architecture tends to be preserved with only minimal distortion, and cytologically atypical nuclei are limited to the basal half of the crypts (Figs. 6 and 7). The nuclei tend to show variable hyperchromasia, overlapping cell borders with nuclear crowding, and irregular nuclear contours. Dystrophic goblet cells may be seen, although typically goblet cell numbers are markedly reduced in dysplastic foci. Separation of LGD from regenerative changes will be discussed below.

Low-magnification view of a focus of low-grade dysplasia in Barrett's esophagus. This focus is recognizable at low magnification because of the pronounced nuclear hyperchromasia.

High-magnification view of low-grade dysplastic glands in Barrett's esophagus. There is variable nuclear hyperchromasia, irregular nuclear contours, and overlapping nuclei.
Simply put, HGD shows more severe cytologic atypia and architectural complexity than is present in LGD, and in some cases this distinction is quite difficult (Fig. 8). Architecturally, there tends to be more crypt complexity in HGD, sometimes with a villiform configuration of the mucosal surface and/or branched or cribriform crypts. Cytologically, the cells show more nuclear pleomorphism and hyperchromatism than is seen in LGD, and there often is nuclear stratification to the crypt luminal surface.

High-magnification view of a focus of high-grade dysplasia in Barrett's esophagus. There is marked cytologic atypia with nuclear stratification to the luminal surface and cytologic atypia that goes beyond that which is seen in low-grade dysplasia.
Separation of intramucosal adenocarcinoma (IMC) from HGD is important, but in some cases, it is exceedingly difficult. By definition, in IMC, neoplastic cells have penetrated through the basement membrane and infiltrate into the lamina propria, typically as single cells or in small clusters. Given the presence of lymphatic channels within the esophageal mucosa, there is a small but definite risk of regional lymph node metastasis in patients with IMC (33, 34). Therapeutic strategies that are based on the histologic separation of HGD from IMC should be looked at with skepticism, given the great difficulty in such histologic separation (35).
A diagnosis of “indefinite for dysplasia,” much to our clinical colleagues' dismay, is a legitimate one. The differentiation of regenerative changes from true dysplasia, particularly in a background of inflammation or ulceration, is at times difficult, if not impossible. Thus, if the pathologist is unsure as to whether the epithelial changes are regenerative or truly dysplastic, a diagnosis of indefinite for dysplasia should be made. In some cases, glandular atypia may be striking in the absence of definitive cytologic atypia in the surface epithelium, and under these circumstances, a diagnosis of indefinite for dysplasia is acceptable as well (described further below).
DISTINGUISHING BETWEEN REGENERATIVE CHANGES AND DYSPLASIA
Because Barrett's mucosa is metaplastic, there is a “baseline atypia” that is always present and in a sense must be overlooked in order to make a diagnosis of dysplasia. This baseline atypia is most pronounced in the glands at the base of the mucosa and does not involve the surface epithelium. In addition, biopsies from Barrett's mucosa are not infrequently inflamed, often with both acute and chronic inflammatory cells. As in the case of active chronic inflammatory bowel disease, neutrophil-mediated epithelial injury can induce regenerative cytologic changes that may be difficult to differentiate from dysplasia. There are some general rules that are useful in distinguishing between these conditions, as outlined below.
One should be conservative about making a diagnosis of dysplasia in the face of active inflammation. Although neutrophils can be found within dysplastic epithelium, the changes have to be convincing in order to make a definitive diagnosis of dysplasia. Otherwise, a diagnosis of indefinite for dysplasia is appropriate unless the changes are clearly regenerative.
The low-magnification appearance of the mucosa is extremely important. True dysplasia usually draws attention at low magnification because of the consistent presence of nuclear hyperchromasia. Obviously, confirmation of cytologic atypia at a higher magnification is necessary. In addition, the cytologic alterations should be present on the surface epithelium, not just in the glandular compartment. In a well-oriented specimen, it is fairly straightforward to determine whether these cytologic alterations involve the surface epithelium. However, in a tangentially sectioned biopsy specimen, this evaluation can be difficult.
Cytologically, dysplastic epithelium tends to show variable nuclear hyperchromasia and pleomorphism. In other words, cells tend to look different from their neighbors, with some showing nuclear hyperchromasia and irregular nuclear contours when compared with surrounding cells within the same crypt. In contrast, although both nuclear hyperchromasia and pleomorphism may be seen in repair, the changes tend to be less severe and more uniform, with cells resembling their neighbors within the same crypt or in adjacent crypts. Thus, the cytologic atypia associated with repair is more uniform than in dysplasia. Dysplastic cells also tend to have a higher nuclear-to-cytoplasmic ratio as well, as irregular nuclear contours. Although regenerative cells may have nuclear size similar to those seen in dysplasia, there tends to be a commensurate increase in the amount of cytoplasm, such that the nuclear-to-cytoplasmic ratio is normal or only mildly increased. In addition, regenerative cells tend to have round and regular nuclear contours.
SAMPLING ERROR AND OBSERVER VARIATION IN THE DIAGNOSIS OF BARRETT'S-RELATED DYSPLASIA
In any given case, dysplasia may be diffusely distributed throughout a BE segment, or the changes may be focal, sometimes limited to a small area of one fragment in a patient with multiple specimens. When dysplasia is diffuse, using the 4-quadrant biopsy technique previously described, there will be a high frequency of detecting the dysplastic foci. However, even using this rigorous sampling technique, small foci of dysplasia can be left unsampled. The need for thorough sampling is further emphasized by the fact that many examples of HGD or early adenocarcinoma arising in BE are not associated with a grossly recognizable lesion (24, 31). Given this sampling error, once a diagnosis of dysplasia is made, subsequent biopsies without dysplasia should not lull the gastroenterologist into a false sense of security, provided that the original diagnosis was correct.
Another problem facing the pathologist and the gastroenterologist (and for that matter, the thoracic surgeon and ultimately the patient) is both the intra- and interobserver variation in the diagnosis of dysplasia. Given the subtle gradation of changes from baseline atypia to LGD to HGD, it is not surprising that this variation exists. Reid et al. (36) found this variation to be most striking at the low end of the histologic spectrum—that is, in distinguishing negative for dysplasia from LGD or indefinite for dysplasia. The study by Reid et al. (36) describes observer variation in terms of percentage agreement, which does not account for agreement that is likely to occur by chance alone. A more recent study by Montgomery et al. (37) using kappa statistical analysis (which does account for agreement that occurs by chance alone) confirmed a high degree of intra- and interobserver variation in the separation of these diagnoses, even among pathologists with a special interest in gastrointestinal pathology.
NATURAL HISTORY OF HIGH-GRADE DYSPLASIA
As previously mentioned, HGD is frequently seen in the mucosa adjacent to invasive esophageal adenocarcinomas and is felt to represent the immediate precursor lesion. However, HGD also is a marker of synchronous or metachronous adenocarcinoma in these patients. For example, in approximately 30 to 40% of esophagi resected for HGD, an unsuspected adenocarcinoma is identified (21, 22, 27, 31, 38). In fact, in our own experience at the Cleveland Clinic Foundation, even with extensive preoperative sampling, patients with a preoperative diagnosis of HGD in the absence of a grossly identifiable lesion still had a deeply invasive carcinoma in the esophagectomy specimen in 33% of cases (31). However, Reid et al. (39) reported excellent success at detecting early carcinomas arising in Barrett's-related HGD in patients who were followed with four-quadrant 1-cm endoscopic biopsy protocols that were performed at closely timed intervals. In a recent prospective study of patients with unifocal HGD with long-term follow-up by Weston et al. (40), 8 (53%) of 15 patients progressed to either invasive carcinoma or multifocal HGD. Those investigators concluded that unifocal HGD has a high risk for progressing to multifocal HGD or invasive carcinoma and discouraged an observational approach when this diagnosis is rendered.
Recently, a large study of patients with BE-related HGD from the Hines VA Hospital (Hines, IL) suggested that surveillance endoscopy with biopsy is a valid and safe follow-up strategy for patients with HGD without cancer (41). Of 1099 patients with BE, 79 (7.2%) initially had HGD, including 34 patients with prevalent HGD and 45 patients who subsequently developed HGD (incident HGD) without evidence of cancer. In 4 of these 79 patients, rigorous endoscopy and biopsy detected an unsuspected adenocarcinoma within the 1st year after detection of HGD (“the hunt”). Of the 75 patients with HGD who remained without detectable cancer after the 1 year of intensive searching, 12 patients (16%) subsequently developed carcinoma during a mean surveillance period of 7.3 years. Eleven of the 12 patients who developed carcinoma were considered cured with surgical or ablation therapy. From these data, those investigators concluded that BE-related HGD without detectable carcinoma follows a relatively benign course in the vast majority of patients. It is interesting that of the 1099 patients with BE, 737 had LGD, a number that far exceeds that of any other previously published study. One experienced gastrointestinal pathologist interpreted all of the biopsy specimens over a 20-year period. Although the results of this study are provocative, additional prospective studies with long-term follow-up in patients with nonsurgical management of HGD are required, a task that may be difficult to reproduce.
MANAGEMENT OF BARRETT'S-RELATED DYSPLASIA
The management of patients with Barrett's-related dysplasia is controversial and varies from institution to institution. Given the paucity of prospective data on the natural history and time course of the dysplasia-carcinoma sequence, there is no standard way to manage these patients. At our institution, the current management plan is a modification of the recommended guidelines proposed by Reid et al. (24) Once a diagnosis of BE is made, patients are followed with yearly endoscopy with biopsy. If a diagnosis of indefinite or LGD is rendered, patients are generally placed on anti-reflux therapy in order to reduce the intensity of inflammation and reactive epithelial changes that could be misinterpreted as dysplasia. After anti-reflux therapy, our gastroenterologists generally repeat endoscopy with biopsy in 3–6 months. If the repeat biopsies are negative, we repeat endoscopy with biopsy at 3–6 month intervals until two consecutive negative interpretations are encountered, followed by a return to yearly surveillance. However, if indefinite or LGD persists, we continue a program of 3–6 month surveillance until dysplasia progresses.
Although the management of HGD is controversial, at our institution, we consider esophagectomy if a diagnosis of HGD is confirmed. Confirmation can be obtained either by review and agreement by an experienced gastrointestinal pathologist or by immediate re-endoscopy and biopsy with a diagnosis of HGD (42). Given our relatively low mortality rate from esophagectomy (around 2%), we believe this procedure is indicated in operative candidates with HGD.
ADJUNCTIVE TECHNIQUES IN SCREENING FOR BARRETT'S-RELATED DYSPLASIA
Several adjunctive techniques have been proposed as having a possible role in the screening for dysplasia in patients with BE, given the limitations and imperfections of routine light microscopy. DNA content, as measured by flow cytometric methods, has been studied in the dysplasia-carcinoma sequence in patients with BE, the results of which have been conflicting. In 1987, Reid et al. (43) found an increased prevalence of DNA aneuploidy and elevated S-phase fraction with increasing severity of the histologic abnormality. In 1992, Reid et al. (23) prospectively studied 62 patients with BE by both histology and flow cytometry. Interestingly, 9 of 13 patients who had aneuploidy or increased G2/tetraploid populations in their initial biopsy specimens developed HGD or carcinoma, with a mean follow-up interval of 34 months. None of the 49 patients without aneuploidy or increased G2/tetraploid populations progressed to HGD or carcinoma. In a more recent prospective study by Reid et al. (44), patients with negative, indefinite, or LGD with neither aneuploidy nor increased 4N fraction had a zero rate of 5-year cumulative cancer incidence, compared with 28% for those with either aneuploidy or increased 4N. Patients with baseline increased 4N, aneuploidy, and HGD had 5-year cancer incidences of 56%, 43% and 59%, respectively. In contrast to the results obtained by Reid et al. (44), Fennerty et al. (45) found discordance between flow cytometric abnormalities and dysplasia in patients with BE. Thus, further studies are necessary to explore the possibility of flow cytometry as a diagnostic adjunct in Barrett's-related dysplasia.
MOLECULAR ALTERATIONS IN THE BARRETT'S METAPLASIA-DYSPLASIA-CARCINOMA SEQUENCE
The colorectal adenoma-carcinoma sequence model has become the paradigm for understanding an accumulation of molecular genetic alterations in a neoplastic process. Over the past several years, numerous studies have evaluated the molecular evolution of the metaplasia-dysplasia-carcinoma sequence in BE, and similar to the case with colorectal adenoma-carcinoma paradigm, there appears to be an accumulation of molecular genetic alterations that are central to the progression of this sequence (46). However, because an inflammatory and metaplastic process forms the basis of the Barrett's-related sequence, at least at the conceptual level, this sequence is more akin to that seen in ulcerative colitis-related dysplasia and carcinoma.
For virtually every marker examined, there is an increased frequency of abnormality of that marker as one progresses along the dysplasia-carcinoma sequence. Certain abnormalities are consistently related to earlier events in this sequence, whereas others appear to be later markers. For example, altered expression of growth factors, including cyclin D1 expression (chromosome 11q13; 47) and hypermethylation or mutation of p16 (chromosome 9p21; 48), seems to be a critical early event. In addition, increased telomerase RNA and protein has been identified not only in early dysplastic lesions but in nondysplastic BE as well (49, 50). In contrast, alterations of p53 (51, 52, 53) and inhibition of apoptosis, possibly mediated by Fas-Fas ligand (54), seem to be later events in this sequence.
Studies similar to the one recently reported by Wu et al. (55) will no doubt continue to be published in the coming years, further refining our understanding of the genetic alterations in the Barrett's-related dysplasia-carcinoma sequence. One of the important potential clinical uses of this knowledge will be defining the “magic bullet” marker that will allow identification of those patients with BE who are truly at risk of progressing along this sequence. Such a marker could allow separation of those patients with BE who are very unlikely to ever have their disease progress and who thus are unlikely to benefit from continued endoscopic surveillance. Until such a marker is found, most would advocate endoscopic surveillance with biopsy in all patients with documented esophageal intestinal metaplasia, a very time- and effort-consuming endeavor, not to mention one with astronomical medical costs involved. The ability to target those patients with Barrett's esophagus who are at much greater risk for progressing to dysplasia or carcinoma would be highly valued by gastroenterologists, pathologists and, most important, patients with long-standing gastroesophageal reflux disease and known intestinal metaplasia.
References
Spechler SJ, Goyal RK . Barrett's esophagus. N Engl J Med 1986; 315: 362–371.
Haggitt RC, Tryzelaar J, Ellis FH, Colcher H . Adenocarcinoma complicating columnar epithelial-lined (Barrett's) esophagus. Am J Clin Pathol 1978; 70: 1–5.
Haggitt RC . Adenocarcinoma in Barrett's esophagus. A new epidemic? Hum Pathol 1992; 23: 475–476.
Blot WJ . Esophageal cancer trends and risk factors. Semin Oncol 1994; 21: 403–410.
Bombeck CT, Dillard DH, Nyhus LM . Muscular anatomy of the gastroesophageal junction and role of phrenoesophageal ligament. Autopsy study of sphincter mechanism. Ann Surg 1966; 164: 643–654.
Der R, Tsao-Wei DD, DeMeester T, Peters J, Groshen S, Lord RV . Carditis. A manifestation of gastroesophageal reflux disease. Am J Surg Pathol 2001; 25: 245–252.
Öberg S, Peters JH, DeMeester TR, Chandrasoma P, Hagen JA, Ireland AP, et al. Inflammation and specialized intestinal metaplasia of cardiac mucosa is a manifestation of gastroesophageal reflux disease. Ann Surg 1997; 226: 522–532.
Chandrasoma PT, Lokuhetty DM, DeMeester TR, Bremner CG, Peters JH, Öberg S, et al. Definition of histopathologic changes in gastroesophageal reflux disease. Am J Surg Pathol 2000; 24: 344–351.
Owen DA . Stomach. In: Sternberg SS, editor. Histology for pathologists. New York: Raven Press; 1992. p. 533–545.
Chandrasoma PT, Der R, Ma Y, Dalton P, Taira M . Histology of the gastroesophageal junction: an autopsy study. Am J Surg Pathol 2000; 24: 402–409.
Kilgore S, Ormsby AH, Gramlich TL, Rice TW, Richter JE, Falk GW, et al. The gastric cardia: fact or fiction? Am J Gastroenterol 2000; 95: 921–924.
Spechler SJ, Goyal RK . The columnar-lined esophagus, intestinal metaplasia and Norman Barrett. Gastroenterology 1996; 110: 614–621.
Paull A, Trier JS, Dalton MD, Camp RC, Loeb P, Goyal RK . The histologic spectrum of Barrett's esophagus. N Engl J Med 1976; 295: 476–480.
Haggitt RC . Barrett's esophagus, dysplasia and adenocarcinoma. Hum Pathol 1994; 25: 982–993.
Weinstein WM, Ippoliti AF . The diagnosis of Barrett's esophagus. Goblets, goblets, goblets. Gastrointest Endosc 1996; 44: 91–94.
Sampliner RE . Practice guidelines on the diagnosis, surveillance and therapy of Barrett's esophagus. Am J Gastroenterol 1998; 93: 1028–1031.
Sharma P, Morales TG, Sampliner RE . Short segment Barrett's esophagus—the need for standardization of the definition and of endoscopic criteria. Am J Gastroenterol 1998; 93: 1033–1036.
Weston AP, Krmpotich P, Makdisi WJ, Cherian R, Dixon A, McGregor DH, et al. Short-segment Barrett's esophagus: clinical and histological features, associated endoscopic findings and association with gastric intestinal metaplasia. Am J Gastroenterol 1996; 91: 981–986.
Offner FA, Lewin KJ, Weinstein WM . Metaplastic columnar cells in Barrett's esophagus: a common and neglected cell type. Hum Pathol 1996; 27: 885–889.
Sjogren RW, Johnson LF . Barrett's esophagus: a review. Am J Med 1983; 74: 3131–3140.
Hamilton SR, Smith RRL . The relationship between columnar epithelial dysplasia and invasive adenocarcinoma arising in Barrett's esophagus. Am J Clin Pathol 1987; 87: 301–312.
Lee RG . Dysplasia in Barrett's esophagus. A clinicopathologic study of six patients. Am J Surg Pathol 1985; 9: 845–852.
Reid BJ, Blount PL, Rubin CE, Levine DS, Haggitt RC, Rabinovitch PS . Predictors of progression to malignancy in Barrett's esophagus: endoscopic, histologic and flow cytometric follow-up of a cohort. Gastroenterology 1992; 102: 1212–1219.
Reid BJ, Weinstein WM, Lewin KJ, Haggitt RC, VanDeventer G, Den Besten L, et al. Endoscopic biopsies diagnose high-grade dysplasia or early operable adenocarcinoma in Barrett's esophagus without grossly recognizable neoplastic lesions. Gastroenterology 1988; 94: 81–90.
Schmidt HG, Riddell RH, Walther B, Skinner DB, Riemann JF . Dysplasia in Barrett's esophagus. J Cancer Res Clin Oncol 1985; 110: 145–152.
Smith RRL, Hamilton SR, Boitnott JK, Rogers EL . The spectrum of carcinoma arising in Barrett's esophagus: a clinicopathologic study of 26 patients. Am J Surg Pathol 1984; 8: 562–573.
Hameeteman W, Tytgat GNJ, Houthoff HJ, van den Tweel JG . Barrett's esophagus: development of dysplasia and adenocarcinoma. Gastroenterology 1989; 96: 1249–1256.
Schnell TG, Sontag SJ, Chejfec G . Adenocarcinomas arising in tongues or short segments of Barrett's esophagus. Dig Dis Sci 1992; 37: 137–143.
Hirota WK, Loughney TN, Lazas DJ, Maydonovitch CL, Rholl V, Wong RK . Specialized intestinal metaplasia, dysplasia and cancer of the esophagus and esophagogastric junction: prevalence and clinical data. Gastroenterology 1999; 116: 277–285.
Menke-Pluymers MBE, Hop WCJ, Dees J, van Blankenstein M, Tilanus HW . Risk factors for the development of an adenocarcinoma in columnar-lined (Barrett) esophagus. Cancer 1993; 72: 1155–1158.
Falk GW, Rice TW, Goldblum JR, Richter JE . Jumbo biopsy forceps protocol still misses unsuspected cancer in Barrett's esophagus with high-grade dysplasia. Gastrointest Endosc 1999; 49: 170–176.
Riddell RH, Goldman H, Ransohoff DF, Appelman HD, Fenoglio CM, Haggitt RC, et al. Dysplasia in inflammatory bowel disease: standardized classification with provisional clinical implications. Hum Pathol 1983; 14: 931–968.
Goseki M, Koike M, Yoshida M . Histopathologic characteristics of early stage esophageal carcinoma. A comparative study with gastric carcinoma. Cancer 1992; 69: 1088–1093.
Sabik JF, Rice TW, Goldblum JR, Koka A, Kirby TJ, Medendorp SV, et al. Superficial esophageal carcinoma. Ann Thorac Surg 1995; 60: 896–902.
Ormsby AH, Petras RE, Henricks WH, Rice TW, Richter JE, Goldblum JR . Observer variation in the diagnosis of superficial adenocarcinoma. Gut 2002; 51: 671–676.
Reid BJ, Haggitt RC, Rubin CE, Roth G, Surawicz CM, Van Belle G, et al. Observer variation in the diagnosis of dysplasia in Barrett's esophagus. Hum Pathol 1988; 19: 166–178.
Montgomery E, Bronner MP, Goldblum JR, Greenson JK, Haber MM, Hart J, et al. Reproducibility of the diagnosis of dysplasia in Barrett's esophagus: a reaffirmation. Hum Pathol 2001; 32: 368–378.
Rice TW, Falk GW, Achkar E, Petras RE . Surgical management of high-grade dysplasia in Barrett's esophagus. Am J Gastroenterol 1993; 88: 1832–1836.
Reid BJ, Blount PL, Feng Z, Levine DS . Optimizing endoscopic biopsy detection of early cancers in Barrett's high-grade dysplasia. Am J Gastroenterol 2000; 95: 3089–3096.
Weston AP, Sharma P, Topalovski M, Richards R, Cherian R, Dixon A . Long-term follow-up of Barrett's high-grade dysplasia. Am J Gastroenterol 2000; 95: 1888–1893.
Schnell TG, Sontag SJ, Chejfec G, Aranha G, Metz A, O'Connell S, et al. Long-term nonsurgical management of Barrett's esophagus with high-grade dysplasia. Gastroenterology 2001; 120: 1607–1619.
Petras RE, Sivak MV, Rice TW . Barrett's esophagus: a review of the pathologist's role in diagnosis and management. Pathol Annu 1991; 26: 1–32.
Reid BJ, Haggitt RC, Rubin CE, Rabinovitch PS . Flow cytometry complements histology in detecting patients at risk for Barrett's adenocarcinoma. Gastroenterology 1987; 93: 1–11.
Reid BJ, Levine DS, Longton G, Blount PL, Rabinovitch PS . Predictors of progression to cancer in Barrett's esophagus: baseline histology and flow cytometry identify low- and high-risk patient subsets. Am J Gastroenterol 2000; 95: 1669–1676.
Fennerty MB, Sampliner RE, Way D, Riddell R, Steinbronn K, Garewal HS . Discordance between flow cytometric abnormalities and dysplasia in Barrett's esophagus. Gastroenterology 1989; 97: 815–820.
Jankowski JA, Wright NA, Meltzer SJ, Triadafilopoulos G, Geboes K, Casson AG, et al. Molecular evolution of the metaplasia-dysplasia-adenocarcinoma sequence in the esophagus. Am J Pathol 1999; 154: 965–973.
Coppola D, Falcone R, Livingston S, Karl R, Nicosia S, Cacho CM . Cyclin D1 expression correlates with degrees of dysplasia in Barrett's esophagus. Lab Invest 1997; 76: 298–302.
Reid BJ, Sanchez CA, Blount PL, Levine DS . Barrett's esophagus: cell cycle abnormalities in advancing stages of neoplastic progression. Gastroenterology 1993; 105: 119–129.
Morales CP, Lee EL, Shay JW . In situ hybridization for the detection of telomerase RNA in the progression from Barrett's esophagus to esophageal adenocarcinoma. Cancer 1998; 83: 652–659.
Mullick T, Tasch JE, Ormsby AH, Richter JE, Goldblum JR, Petras RE, et al. Telomerase upregulation in Barrett's esophagus precedes the development of esophageal adenocarcinoma. Am J Gastroenterol 2001; 96: 75A.
Casson AG, Mukhopadhay T, Cleary KR, Ro JY, Levin B, Roth JA . p53 gene mutations in Barrett's epithelium and esophageal cancer. Cancer Res 1991; 51: 4495–4499.
Younes M, Lebovitz RM, Lechago LV, Lechago J . p53 protein accumulation in Barrett's metaplasia, dysplasia and carcinoma: a follow-up study. Gastroenterology 1993; 105: 1637–1642.
Haamelin R, Flejou JF, Muzeau F, Potet F, Laurent-Puig P, Fekete F, et al. TP53 mutations, and p53 protein immunoreactivity in malignant and premalignant Barrett's oesophagus. Gastroenterology 1994; 107: 1012–1018.
Younes M, Schwartz MR, Finnier D, Younes A . Overexpression of Fas ligand (FasL) during malignant transformation in the large bowel and in Barrett's metaplasia of the esophagus. Hum Pathol 1999; 30: 1309–1313.
Wu T-T, Watanabe T, Heitmiller R, Zahurak M, Forastiere AA, Hamilton SR . Genetic alterations in Barrett esophagus and adenocarcinomas of the esophagus and esophagogastric junction region. Am J Pathol 1998; 153: 287–294.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Goldblum, J. Barrett's Esophagus and Barrett's-Related Dysplasia. Mod Pathol 16, 316–324 (2003). https://doi.org/10.1097/01.MP.0000062996.66432.12
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.MP.0000062996.66432.12
Keywords
This article is cited by
-
Forkhead box F1 induces columnar phenotype and epithelial-to-mesenchymal transition in esophageal squamous cells to initiate Barrett's like metaplasia
Laboratory Investigation (2021)
-
Barrett oesophagus: lessons on its origins from the lesion itself
Nature Reviews Gastroenterology & Hepatology (2015)
-
Treatment of High-Grade Dysplasia and Early Stage Esophageal Adenocarcinoma with an Endoscope: The Ultimate in Minimally Invasive, Curative Therapy
Current Surgery Reports (2014)
-
Comparison of six immunohistochemical markers for the histologic diagnosis of neoplasia in Barrett’s esophagus
Virchows Archiv (2010)
-
Dendritic Cell-Associated Immune Inflammation of Cardiac Mucosa: A Possible Factor in the Formation of Barrett’s Esophagus
Journal of Gastrointestinal Surgery (2009)
