
Abstract
Pleomorphic liposarcoma is a variant of liposarcoma defined morphologically by the presence of pleomorphic lipoblasts. Because of its rarity, there are limited studies with long-term follow-up information. Nineteen pleomorphic liposarcomas were studied. Unequivocal pleomorphic lipoblasts were required for inclusion. In each case, the following features were noted: tumor site; tumor size; tumor depth; predominant histologic pattern (epithelioid or malignant fibrous histiocytoma [MFH]-like); extent of necrosis (absent, less than 15%, or at least 15%); mitotic counts; treatment and clinical follow-up. Patients were 11 females and 8 males, aged 33–87 years (mean, 64.5 y; median, 70 y). Tumors involved the extremities (13 patients: intramuscular in 10, subcutaneous in 2, depth unknown in 1), retroperitoneum (4 patients), mediastinum (1 patient), and paratesticular region (1 patient). Size ranged from 4.5–31 cm (mean, 11.9 cm; median, 12.0 cm). Predominant pattern was epithelioid in 7 and MFH-like in 12. Necrosis was present in 15 (79%) and was extensive (36 15%) in 14 patients. Mitotic counts ranged from 0.2–3.4/10 high-power fields (mean, 1.4; median, 1.4) by the average-count method and from 1–6/10 high power fields by the highest count method (mean, 2.9; median, 3.0). All patients were treated surgically; 10 patients received adjuvant chemotherapy and/or radiation therapy. On follow-up of 18 patients (range, 2–129 mo; mean, 35.4 mo; median, 23 mo) nine (50%) were dead of disease (range, 2–48 mo; mean, 20.1 mo; median, 12 mo), one died of other causes 2 months after diagnosis, two were alive with disease, five were disease free, and one was alive at 129 months (tumor status unknown). Five had recurrences (range, 3–28 mo; mean, 14.4 mo; median, 8 mo), and four of five (80%) with recurrences were dead of disease. Metastases developed in eight patients (range, 4–48 mo; mean, 19.5 mo; median, 11.5 mo), most commonly to the lungs. In conclusion, pleomorphic liposarcoma is a rare tumor of adulthood that occurs most commonly in the deep, soft tissues of the extremities. It behaves as a high-grade sarcoma that frequently metastasizes, most commonly to the lungs. Although this tumor has a wide range of histologic appearances, no clinical or pathologic feature is predictive of a more aggressive clinical course.
Similar content being viewed by others


INTRODUCTION
Liposarcomas are malignant neoplasms that display light-microscopic evidence of fatty differentiation in the form of lipoblasts. This group of tumors can be conceptualized into three main categories on the basis of distinct clinicopathologic and cytogenetic features: well-differentiated/dedifferentiated liposarcoma, myxoid/round cell liposarcoma, and pleomorphic liposarcoma (PLS; 1, 2).
PLS is the rarest subtype of liposarcoma and is discriminated from other high-grade sarcomas by the presence of pleomorphic lipoblasts (3). Because of the rarity of this tumor, there are few large studies of PLS with long-term follow-up information. We present an analysis of 19 patients with PLS who were accumulated over a 24-year period from three different institutions. The purpose of this study was to analyze the clinical and pathologic features of this rare tumor to determine clinical outcome and to evaluate which parameters, if any, are predictive of a more aggressive clinical course.
MATERIALS AND METHODS
The authors' consultation files and the surgical pathology files of the Cleveland Clinic Foundation, Georgetown University Hospital, and the Royal Marsden NHS Trust were searched for patients diagnosed as pleomorphic liposarcoma, bizarre liposarcoma, and atypical liposarcoma for which hematoxylin and eosin–stained slides were available for review. Pathology reports were reviewed to assess sampling and maximum tumor size. Patients without evidence of lipomatous differentiation in the form of unequivocal pleomorphic lipoblasts, defined as pleomorphic cells containing sharply defined lipid droplets that indent or scallop the nucleus, were excluded, as were inadequately sampled tumors and those patients with inadequate hematoxylin and eosin–stained slides for review. Nineteen patients qualified for the study. Clinical data and follow-up information were obtained by review of medical records and by correspondence with referring pathologists and clinicians.
All slides in each case were reviewed, and the following features were recorded: tumor site; tumor size; tumor depth; predominant histologic pattern, characterized as either epithelioid or malignant fibrous histiocytoma (MFH)-like; extent of tumor cell necrosis (absent, less than 15%, at least 15%); mitotic counts, counting in the most mitotically active area of each tumor using the highest count and average count methods, as previously described (4); treatment; and clinical follow-up.
RESULTS
Clinical Features
The tumors occurred in 11 women and eight men, who had a mean age of 64.5 years (range, 33–87 y; median, 70 y). Thirteen tumors were located in the extremities with 10 intramuscular, 2 subcutaneous, and 1 with an unknown depth. Four tumors were in the retroperitoneum and one in the mediastinum; one was paratesticular. All patients were treated surgically, and 10 patients received adjuvant chemotherapy and/or radiation. See Table 1.
Pathologic Features
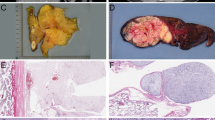
Grossly, the tumors were typically described as firm, nodular masses that were white to yellow on sectioning (Fig. 1). Some of the cases had a myxoid component, and most had visible areas of necrosis. Although the size of the tumors ranged from 4.5 to 31 cm, most were large (more than 10 cm in diameter), with a mean diameter of 11.9 cm and a median of 12 cm. See Table 1.

Gross appearance of a pleomorphic liposarcoma arising in the deep soft tissues of the upper arm of an 87-year-old male.
Histologically, seven tumors were composed predominantly of epithelioid cells with variable numbers of lipoblasts (Fig. 2). The epithelioid cells were generally arranged in large sheets and had round nuclei, often prominent nucleoli, and variably eosinophilic to vacuolated cytoplasm. Scattered pleomorphic epithelioid cells were also seen. Twelve of the tumors were composed predominantly of highly cellular and pleomorphic spindled cells resembling MFH, with variable numbers of lipoblasts (Figs. 3, 4). In these cases, there was marked cellular pleomorphism, including large multinucleated giant cells and scattered inflammatory cells. The pleomorphic lipoblasts varied in number from case to case and between different areas within the same neoplasm. In some cases, only scattered individual or small collections of lipoblasts were seen, but in most, larger collections or nodules of lipoblasts were easily identified. Necrosis was present in 15 cases (79%) and was extensive (at least 15% of the tumor) in 14 cases. Mitotic counts ranged from 0.2 to 3.4/10 high-power fields (HPF; mean, 1.4; median, 1.4) by the average-count method and from 1 to 6/10 HPF (mean, 2.9; median, 3.0) by the highest-count method. Over 70% of the tumors had two or fewer mitotic figures per 10 HPF, as determined by the average mitotic count method.

High-magnification view of a pleomorphic lipoblast in a tumor composed predominantly of cells with an epithelioid morphology.

MFH-like area of a pleomorphic liposarcoma. Pleomorphic lipoblasts were only focally present in this tumor. In their absence, the tumor likely would have been categorized as an MFH.

Area of lipoblastic differentiation in an MFH-like area of a pleomorphic liposarcoma.
Clinical Follow-Up
Follow-up information was obtained in 18 cases. The follow-up interval ranged from 2 to 129 months (mean, 35.4 mo; median, 23 mo). Nine patients (50%) were dead of disease within 4 years of diagnosis (range, 2–48 mo; mean, 20.7 mo; median, 12 mo). One patient died from renal failure 2 months after diagnosis. Two patients were alive with metastatic disease at 6 and 87 months. Five patients were disease free from 6–119 months, and one was alive at 129 months with tumor status unknown. Five patients had recurrences between 3 and 28 months after initial excision, with a mean time to recurrence of 14 months. All but one patient with recurrence died of disease. Eight patients developed metastases between 4 and 48 months after initial diagnosis, with a mean time to metastasis of 19.5 months. The most common site of metastasis was the lung (7 cases), but one patient developed liver metastases. No histologic feature appeared to be predictive of clinical outcome. See Table 2.
DISCUSSION
There are essentially three major subtypes of liposarcoma, each of which is characterized by its own clinicopathologic and cytogenetic features. Well-differentiated/dedifferentiated liposarcoma can be considered to lie within the same spectrum because dedifferentiated liposarcoma represents progression from low-grade, well-differentiated liposarcoma (5, 6). Both are characterized cytogenetically by ring or giant marker chromosomes derived from 12q13–15 (7, 8), although additional cytogenetic aberrations are often detected in dedifferentiated variants (9). Similarly, myxoid/round-cell liposarcoma can be considered along a histologic spectrum; round-cell liposarcoma is the poorly differentiated form of myxoid liposarcoma (10), and they share the characteristic t(12;16) (11). PLS is a clinically, histologically, and cytogenetically distinct form of liposarcoma and is the rarest subtype. Because it is so uncommon and is often difficult to differentiate from other high-grade sarcomas, there are limited data in the literature detailing its clinical and pathologic spectrum. The rarity of this neoplasm is exemplified by the fact that only 19 well-documented cases could be culled from three relatively large institutions over a 24-year period. Similarly, in a study of 66 liposarcomas by Evans (12), only three (4.5%) were of the pleomorphic type.
In general, recognition of PLS requires careful examination of hematoxylin and eosin–stained slides for the presence of pleomorphic lipoblasts; immunohistochemistry does not play a significant role in the recognition of this neoplasm. It is well recognized that lipoblasts are “in the eye of the beholder,” and although the phenomenon has not formally studied, there clearly is some degree of interobserver variation in the identification of lipoblasts (1). This is particularly true when a high-grade spindled or epithelioid neoplasm infiltrates fat, which may result in the isolation of single or small groups of adipocytes that resemble lipoblasts. Despite these difficulties, we believe our stringent criteria for the recognition of pleomorphic lipoblasts have allowed us to isolate a discrete group of neoplasms.
In the majority of cases, PLS resembles an MFH-like tumor, characterized by a predominance of pleomorphic spindled cells with occasional multinucleated giant cells, often arranged in a storiform growth pattern. The difficulty in separating PLS from other high-grade pleomorphic sarcomas has been emphasized by Fletcher (13) in his review of 159 such tumors, 34 (21%) of which were reclassified as PLS on review. Given that some of our examples of PLS had only focal collections of lipoblasts, adequate sampling of the tumor is necessary for recognition of lipoblastic differentiation when it is present.
A smaller subgroup of PLS is composed predominantly of epithelioid cells. This subtype, as recently delineated by Miettinen and Enzinger (14), is composed of sheets of epithelioid cells with round nuclei, variably prominent nucleoli, and cytoplasm that ranges from eosinophilic to vacuolated. In some tumors, the cells are arranged around a distinctly hemangiopericytoma-like vasculature. In the present series, seven tumors were of this epithelioid type; in all of these cases, scattered pleomorphic lipoblasts were found.
The cases of PLS in our study displayed a wide range of mitotic activity and necrosis. Although some tumors had very little necrosis, the vast majority showed extensive areas of necrosis, composing at least 15% of the tumor. Interestingly, over 70% of the tumors in this study had two or fewer mitoses per 10 HPF. Thus, despite the fact that these tumors were typically highly cellular, pleomorphic, and often had extensive areas of necrosis, mitotic figures were relatively inconspicuous in many cases.
PLS is generally considered to be a high-grade sarcoma, given its high rate of recurrence and metastasis. In the study by Enzinger and Winslow published in 1962 (15), 73% of patients with PLS developed a local recurrence, with only 21% surviving 3 years. Subsequent studies of liposarcoma that included examples of PLS showed similar rates of recurrence, metastasis, and survival (16, 17, 18). In a preliminary report of 31 cases of PLS by Mentzel et al. (19), 5 (21%) of 24 patients with follow-up information had a local recurrence, and 7 (29%) developed metastatic disease. Overall, 38% of patients died of their tumors; all had deep lesions.
Our findings confirm the results of these prior studies in that recurrence, metastasis, and death due to tumor occurred in 28%, 44% and 50% of patients, respectively. All but one patient who had recurrence died of disease, and as of last follow-up, all but one patient with metastatic disease also had died. The mean follow-up period in this study was only 35.4 months, and it is likely that survival rates would have been even worse with longer follow-up. As with most sarcomas, the lung is the most common site of metastasis. We were unable to identify any clinical or pathologic feature that was predictive of a more aggressive clinical course.
The differential diagnosis of PLS is broad and depends upon whether an epithelioid or MFH-like pattern predominates. The epithelioid variant may be mistaken for a carcinoma, particularly renal cell and adrenal cortical carcinoma. This difficulty is compounded by the fact that up to 50% of epithelioid PLS show focal immunoreactivity for cytokeratins (14). Fortunately, epithelioid PLS does not express epithelial membrane antigen, a helpful feature in distinguishing this tumor from renal cell carcinoma, which is typically positive for epithelial membrane antigen. Furthermore, the cells of epithelioid PLS have large lipid-filled vacuoles, whereas those in renal cell carcinoma show multiple fine intracytoplasmic lipid droplets. The distinction from adrenal cortical carcinoma may be similarly difficult. The latter tumor typically lacks both cytokeratin and epithelial membrane antigen. In addition, the cells of adrenal cortical carcinoma have fine lipid droplets as opposed to large lipid-filled vacuoles. Other sarcomas with epithelioid features may also enter the differential diagnosis, including epithelioid sarcoma and epithelioid leiomyosarcoma, but a panel of immunohistochemical stains including cytokeratin, epithelial membrane antigen, CD34, and muscle markers will allow for accurate separation of these entities.
The majority of PLS resemble other high-grade pleomorphic sarcomas, the differential diagnosis of which includes pleomorphic leiomyosarcoma, pleomorphic rhabdomyosarcoma, pleomorphic malignant peripheral nerve sheath tumor, and MFH. As emphasized by Fletcher (13), separation of these various types of pleomorphic sarcomas requires careful attention to light-microscopic detail. Although a panel of immunohistochemical markers and even ultrastructural evaluation may be useful in distinguishing between these various pleomorphic sarcomas, the prognostic and therapeutic utility of separating these entities has yet to be fully investigated. Regardless, the recognition of pleomorphic lipoblasts is the sine qua non for identification of PLS; immunohistochemical stains are more helpful in excluding other types of pleomorphic sarcomas than confirming a diagnosis of PLS.
The single benign lesion that could be mistaken for PLS is pleomorphic lipoma, but the latter typically arises as a well-circumscribed subcutaneous mass in the posterior neck, upper shoulders or back of middle-aged males (20). Histologically, pleomorphic lipoma is characterized by an admixture of adipocytes and atypical hyperchromatic cells, including multinucleated floret-like giant cells, as well as characteristic ropey collagen bundles. Although lipoblasts may rarely be seen in pleomorphic lipoma, pleomorphic lipoblasts are not seen. Immunohistochemically, pleomorphic lipomas are characteristically strongly immunoreactive for CD34 (21).
In conclusion, PLS is a rare tumor of adulthood that occurs most commonly in the deep soft tissues of the extremities. It behaves as a high-grade sarcoma that frequently metastasizes, most commonly to the lungs. Histologically, most examples of PLS are MFH-like, differing by the presence of pleomorphic lipoblasts, although a smaller subset is composed predominantly of a proliferation of epithelioid cells. This tumor has a wide range of histologic appearances, but no clinical or pathologic feature appears to be predictive of a more aggressive clinical course.
References
Mentzel T, Fletcher CDM . Lipomatous tumours of soft tissues: an update. Virchows Arch 1995; 427: 353–363.
Dei Tos AP, Dal Cin P . The role of cytogenetics in the classification of soft tissue tumors. Virchows Arch 1997; 431: 83–94.
Enzinger FM, Weiss SW . Liposarcoma. In: Soft tissue tumors. 3rd ed. St Louis, MO: Mosby; 1995. p. 431–466.
Prayson RA, Hart WR . Mitotically active leiomyomas of the uterus. Am J Clin Pathol 1992; 97: 14–20.
Weiss SW, Rao VK . Well-differentiated liposarcoma (atypical lipomatous tumors) of deep soft tissue of the extremities, retroperitoneum, and miscellaneous sites: a follow-up study of 92 cases with analysis of the incidence of dedifferentiation. Am J Surg Pathol 1992; 16: 1051–1058.
Henricks WH, Chu YC, Goldblum JR, Weiss SW . Dedifferentiated liposarcoma: a clinicopathologic analysis of 155 cases with a proposal for an expanded definition of dedifferentiation. Am J Surg Pathol 1991; 21: 271–281.
Fletcher CDM, Akerman M, Dal Cin P, De Wever I, Mandahl N, Mertens F, et al. Correlation between clinicopathologic features and karyotype in lipomatous tumors. A report of 177 cases from the Chromosome and Morphology (CHAMP) Collaborative Study Group. Am J Pathol 1996; 148: 623–630.
Rosai J, Akerman M, Dal Cin P, De Wever I, Fletcher CDM, Mandahl N, et al. Combined morphologic and karyotypic study of 59 atypical lipomatous tumors. Evaluation of their relationship and differential diagnosis with other adipose tissue tumors (a report of the CHAMP study group). Am J Surg Pathol 1996; 20: 1182–1189.
Mertens F, Fletcher CDM, Dal Cin P, De Wever I, Mandahl N, Mitelman F, et al. Cytogenetic analysis of 46 pleomorphic soft tissue sarcomas and correlation with morphologic and clinical features: a report of the CHAMP study group. Genes Chromosomes Cancer 1998; 22: 16–25.
Smith TA, Easley KA, Goldblum JR . Myxoid/round cell liposarcoma of the extremities: a clinicopathologic study of 29 cases with particular attention to extent of round cell liposarcoma. Am J Surg Pathol 1996; 20: 171–180.
Tallini G, Akerman M, Dal Cin P, De Wever I, Fletcher CDM, Mandahl N, et al. Combined morphologic and karyotypic study of 28 myxoid liposarcomas: implications for a revised morphologic typing (a report from the CHAMP group). Am J Surg Pathol 1996; 20: 1047–1055.
Evans HL . Liposarcomas and atypical lipomatous tumours: a study of 66 cases followed for a minimum of 10 years. Surg Pathol 1988; 1: 41–54.
Fletcher CDM . Pleomorphic malignant fibrous histiocytoma: fact or fiction? A critical reappraisal based on 159 tumors diagnosed as pleomorphic sarcoma. Am J Surg Pathol 1992; 16: 213–228.
Miettinen M, Enzinger FM . Epithelioid variant of pleomorphic liposarcoma: a study of 12 cases of a distinctive variant of high-grade liposarcoma. Mod Pathol 1999; 12: 722–728.
Enzinger FM, Winslow DJ . Liposarcoma: a study of 103 cases. Virchows Arch Pathol Anat 1962; 335: 367–388.
Kindblom LG, Angervall L, Svendsen P . Liposarcoma: a clinicopathologic, radiographic and prognostic study. Acta Pathol Microbiol Scand A 1975; 253 (Suppl): 1–71.
Azumi N, Curtis J, Kempson RL, Hendrickson MR . Atypical and malignant neoplasms showing lipomatous differentiation: a study of 11 cases. Am J Surg Pathol 1987; 11: 161–183.
Chang HA, Hajdu SI, Collin C, Brennan MF . The prognostic value of histologic subtypes in primary extremity liposarcoma. Cancer 1989; 64: 1514–1520.
Mentzel T, Bosenberg M, Fletcher CDM . Pleomorphic liposarcoma: clinicopathologic and prognostic analysis of 31 cases. Mod Pathol 1999; 12: 55A.
Shmookler BM, Enzinger FM . Pleomorphic lipoma: a benign tumor simulating liposarcoma. A clinicopathologic analysis of 48 cases. Cancer 1981; 47: 126–133.
Suster S, Fisher C . Immunoreactivity for the human hematopoietic progenitor cell antigen (CD34) in lipomatous tumors. Am J Surg Pathol 1998; 21: 195–200.
Acknowledgements
The authors thank Kathleen Ranney for her assistance in the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Downes, K., Goldblum, J., Montgomery, E. et al. Pleomorphic Liposarcoma: A Clinicopathologic Analysis Of 19 Cases. Mod Pathol 14, 179–184 (2001). https://doi.org/10.1038/modpathol.3880280
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880280
Keywords
This article is cited by
-
Primary pulmonary pleomorphic liposarcoma found with a massive hemothorax
General Thoracic and Cardiovascular Surgery Cases (2023)
-
Systemic Therapy in Advanced Pleomorphic Liposarcoma: a Comprehensive Review
Current Treatment Options in Oncology (2023)
-
Survivorship and prognostic factors for pleomorphic liposarcoma: a population-based study
Journal of Orthopaedic Surgery and Research (2021)
-
Primary anatomical site as a prognostic factor for pleomorphic liposarcoma
Journal of Cancer Research and Clinical Oncology (2020)
-
Successful excision of large mediastinal liposarcoma with bilateral thoracic extension
Indian Journal of Thoracic and Cardiovascular Surgery (2018)
