
Abstract
Primary non-Hodgkin's lymphomas of bone (PNHLB) is a rare form of extranodal lymphoma. Many studies have reported the clinical, radiologic, and histopathologic characteristics of PNHLB; however, their molecular features have not been well studied. In this report, we present the immunophenotypic and molecular characteristics of 20 primary large B-cell lymphoma (PLBCL) of bone from 20 adults. Most demonstrated centroblastic morphology, with the majority exhibiting nuclear multilobation. One case (5%) demonstrated anaplastic features with strong CD30 expression but was ALK-1 negative. BCL-6 expression was seen in 6 of 20 cases, and strong p53 protein expression was seen in 11 of 20 (55%) cases. The majority of cases analyzed (13/18 = 72%) demonstrated a clonal B-cell process by IgH gene rearrangement studies. Of the five cases that did not demonstrate a clonal population, two expressed BCL-6 protein. No cases demonstrated a bcl-2/JH rearrangement, but BCL-2 protein expression was seen in 11 of 20 (55%) cases. In summary, primary lymphoma of bone is largely a non-Hodgkin's lymphoma of large B-cell type. Our studies demonstrate that p53 and BCL-2 expression may play a role in the pathogenesis of PLCBL of bone. In addition, a subset of the cases are of putative germinal center B-cell origin based on the expression of BCL-6 protein and may be genetically distinct from follicle center lymphomas. The results provide evidence for molecular heterogeneity within primary large B-cell lymphomas of bone.
Similar content being viewed by others


INTRODUCTION
Primary non-Hodgkin's lymphoma of bone (PNHLB) is a rare disease comprising approximately 4 to 5% of extranodal lymphomas (1, 2) and <1% of all non-Hodgkin's lymphomas (1). Clinically defined as “lymphoma presenting in an osseous site with no evidence of disease elsewhere for at least six months after diagnosis,” it was first established as a clinical entity in 1939 (3). The majority of PNHLB have been more recently characterized as diffuse large B-cell lymphomas (DLBCL) using the REAL classification (4, 5). Although many studies have reported the clinical, radiologic, and histopathologic parameters of PNHLB, few have addressed the molecular features to any significant extent (1, 4, 6, 7, 8, 9, 10), likely because of their rarity and the paucity of tissue available in small biopsy specimens. Only a single case report of PNHLB has included molecular studies to detect clonal immunoglobulin heavy-chain (IgH) gene rearrangements (11), whereas another describes other common gene rearrangements found in DLBCL (12). Immunophenotyping of PNHLB with either immunohistochemistry or flow cytometry has been performed, but few of these studies have included evaluation of BCL-2 or p53 protein expression, which have been implicated as prognostic indicators in nodal and extranodal DLBCL (13, 14). The histogenetic derivation of primary bone lymphomas is unknown, and the analysis of germinal-center B-cell–associated expression of BCL-6 (15) and other markers used to subclassify DLBCLs has not been reported in PNHLB. Moreover, occurrence of anaplastic large-cell lymphoma (ALCL) of either T-/null- or B-cell phenotype has been reported (5, 16, 17, 18); however, the incidence of ALK-1 expression has been studied in only a subset of those studies (5, 18).
In this report, we describe 20 cases of primary large B-cell lymphoma (PLBCL) of bone diagnosed and treated at a single institution over a 3-year interval. Molecular analysis demonstrated clonal rearrangement of the immunoglobulin heavy-chain gene in 72% of cases without evidence of a bcl-2/JH gene rearrangement.
MATERIALS AND METHODS
Cases
Paraffin blocks and clinical histories from 23 patients presenting with primary bone and associated soft-tissue lesions were retrieved from the archives of Mount Sinai Hospital, Toronto, Ontario, Canada, a referral center for patients with bone tumors. All cases were diagnosed and treated at Mount Sinai Hospital during the period between 1995 and 1998. Only cases fulfilling the criteria for diagnosis of primary lymphoma of bone (bone lesion ± local soft tissue component as primary site of presentation without evidence of disease elsewhere within a 6-month period) were included in the study. Clinical features including sex, age, and anatomic location were evaluated.
Histologic and Immunophenotypic Studies
Histologic sections were prepared from 10% formalin-fixed or B5-fixed, paraffin-embedded tissues. All sections were stained with hematoxylin and eosin and studied by light microscopy. Immunohistochemical stains were performed on formalin-fixed, paraffin-embedded tissue using the avidin-biotin technique with diaminobenzidine as a chromogen and standard manual methods or an automated immunostainer (DAKO Stainer, Missisauga, Ontario, Canada) (19). Tissue sections were stained with monoclonal antibodies: CD45, CD20, CD43, CD30, BCL-2, p53, BCL-6, ALK-1, and MIB-1 (Ki-67). Appropriate positive and negative controls were performed for all antibodies. The source of antibodies used was as follows: anti-CD20 (L26, Becton Dickinson, Mountain View, CA); anti-CD45 (DAKO); anti-CD43 (DFT1, DAKO); anti-CD30 (BerH2, DAKO); anti-BCL-2 (124, DAKO); anti-p53 (D07, Novocastra, Burlington, Ontario); anti–BCL-6 (DAKO); anti-ALK-1 (DAKO); and anti-MIBI (Ki-67, DAKO).
Interpretation
Lymphomas were classified using morphologic and immunohistochemical criteria described in the Revised European-American Lymphoma Classification (20). All cases were independently assessed by three hematopathologists (MSL, DH-C, BF). Immunohistochemical studies were evaluated as follows. For CD20, BCL-6, BCL-2, and CD30: (−), no staining; (+), 0 to 50% positive; and (++), >50% positive. For p53: (+), 0 to 25% positive; (++), 26 to 50% positive; and (+++), >50% positive. For MIB1: (++), 0 to 50% positive and (+++), >50% positive. A T-cell lymphoblastic lymphoma, a B-cell small lymphocytic lymphoma, and an anaplastic myeloma were excluded from the subsequent studies because long-term clinical followup was not available to confirm their primary bone origin.
Molecular Studies
Genomic DNA was extracted from 10-μm-thick paraffin-embedded sections using standard protocol (21). Sections of paraffin-embedded tissue were deparaffinized with xylene for 30 minutes, then dehydrated with 100% and 70% ethanol for 20 minutes each, followed by washing with dH20 twice and digested with proteinase K at 42° C overnight. The digested DNA solution was then boiled for approximately 10 minutes to inactivate proteinase K. Immunoglobulin heavy-chain gene (IgH) rearrangements were evaluated by polymerase chain reaction (PCR) using VH region Framework 3 (Fr3A) consensus primer and a JH consensus primer as reported elsewhere (22). Primer sequences for IgH-PCR were JH (5′-ACC TGA GGA GAC GGT GAC C-3′) and Fr3A (5′-ACA CGG CTA TGT ATT ACT GT-3′). The PCR reaction mixture consisted of 0.5 μg (approximately 30 pmol) of each oligonucleotide primer, 1 μL of sample DNA (1/50 dilution of the DNA digestion solution), 1 unit of Taq Polymerase Gold, 2.5 mm Tris (pH 8.3), 2.5 mm KCl, 3 mm MgCl2, and 0.5 mm of dNTP Mix (PerkinElmer Life Sciences, Inc., Boston, MA) at a total volume of 25 μL. The PCR protocol consisted of an initial denaturation step of the reaction mixture at 96° C for 10 minutes, followed by 40 cycles of denaturation at 96° C for 30 seconds, annealing of the primer at 60° C for 30 seconds, and extension of the DNA at 72° C for 30 seconds. Final extension of the DNA was performed at 72° C for 5 minutes.
Analysis of bcl-2/IgH gene rearrangement was assessed at the major breakpoint region (MBR) and the minor cluster region (MCR) as described elsewhere (23, 24). Amplification of DNA flanking the bcl-2/JH breakpoint was performed by PCR using a consensus JH (5′-ACC TGA GGA GAC GGT GAC C-3′) and MCR (5′-GAC TCC TTT ACG TGC TGG TAC CC-3′) and MBR (5′-ACC TGA GGA GAC GGT GAC CAG GGT-3′). The amplification conditions for MBR/JH and MCR/JH PCR were as follows: an initial 96° C for 10 minutes, followed by 40 cycles at 96° C for 0.5 minutes, at 60° C for 30 seconds, and at 72° C for 30 seconds, followed by final extension at 72° C for 5 minutes. The reaction contained 0.5 μg (approximately 30 pmol) of each oligonucleotide primer, 1 μL of sample DNA (1/50 dilution of the DNA digestion solution), 1 unit of Taq Polymerase Gold, 2.5 mm KCl, 3 mm MgCl2, and 0.5 mm of a dNTP Mix (Perkin-Elmer) at a total volume of 25 μL. The amplified PCR product for MBR is 600–620 bps, and for MCR, 500–560 bps.
The PCR products were electrophoresed in 1.5% Synergel (Diversified Biotech, Boston, MA) containing 0.5 μg ethidium bromide per milliliter and photographed under ultraviolet light. Appropriate precautions were used to prevent contamination of PCR reactions, including physical separation of the PCR setup from product analysis. The negative control in each amplification experiment was DNA from human placenta, and the positive control was the DNA from previously confirmed positive case.
RESULTS
Clinical Features
The clinical features of the 20 patients are summarized in Table 1. Patients ranged in age from 25 to 86 years (mean, 56.5 y), with 17 males and 3 females (male–female ratio of 5.7:1). The sites of involvement included femur (4 cases), humerus (8 cases), pelvis (6 cases), and miscellaneous (4 cases). Multifocal bone involvement was noted in two cases. Patients 1 and 7 presented with recurrences involving different bone sites.
Histologic and Immunohistochemical Features
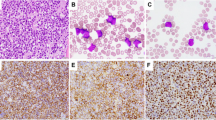
The histologic and immunophenotypic characteristics of the 20 large B-cell lymphoma cases are summarized in Table 1 and presented in Figures 1 to 2. The remaining cases consisting of a precursor T-lymphoblastic lymphoma, a B-cell small lymphocytic lymphoma/chronic lymphocytic lymphoma, and an anaplastic myeloma were excluded from further studies. Eighteen of the large B-cell lymphoma cases demonstrated centroblastic morphology with multilobated nuclei (Figs. 1A and 2A) in 13 (75%) cases, whereas one (5%) demonstrated immunoblastic morphology with round nuclei, abundant amphophilic cytoplasm, and single prominent eosinophilic nucleoli (Fig. 1B). One case demonstrated anaplastic features with a prominent neutrophilic infiltrate (Fig. 1C) with multilobulated nuclei and abundant granular cytoplasm. There was diffuse strong immunoreactivity for CD30 (Fig. 1D). CD15 and ALK-1 were negative in this tumor, and it was considered to be a DLBCL with anaplastic features. The tumor expressed BCL-2 but was BCL-6 negative. All cases expressed CD20 or CD79a (data not shown).

A lymphoma exhibiting multilobated nuclear features with three to four small nucleoli (A; hematoxylin and eosin [H & E], 400 ×). A case with immunoblastic morphology with round nuclei, and prominent eosinophilic nucleoli (B; H & E, 400 ×). A lymphoma with anaplastic morphology is surrounded by a neutrophil-rich infiltrate (C; H & E, 400 ×). The tumor expresses strong membranous and Golgi pattern of CD30. (D; 200 ×).

Primary non-Hodgkin's lymphoma of bone with centroblastic morphology with significant nuclear pleomorphism, finely clumped chromatin, and two to three distinct nucleoli. Numerous mitotic figures and karyorrhectic debris are present. (A; hematoxylin and eosin staining, 400 ×) The tumor cells express nuclear BCL-6 (B; 600 ×), BCL-2 (C; 400 ×), and p53 (D; 200 ×).
Within the large B-cell lymphomas, strong diffuse nuclear expression of BCL-6 (Fig. 2B) was seen in >50% (++) of the tumor cells in 6 of 20 (25%) cases. Strong (>50%; ++ or +++) BCL-2 (Fig. 2C) expression was seen in 11 of 20 (55%) LBCLs. There was moderate to strong (++ and +++) expression of p53 (Fig. 2D) in 11 of 20 (55%) LBCLs. Only 1 of 20 cases (Case 9) was p53 negative. All BCL-2–positive cases showed p53 protein expression. Four of five BCL-6–positive lymphomas also expressed BCL-2 and p53. MIB-1 (Ki-67) yielded satisfactory results in 15 of 20 (75%) cases with ++ to +++ staining in all 15 cases demonstrative of moderate proliferative indices.
Cases 1 and 7 had recurrent lesions at different bone sites. Patient 1 initially presented with DLBCL with centroblastic cytology. The tumor was negative for BCL-2 and BCL-6 but expressed p53. The recurrence that occurred 1 year later was positive for both BCL-2 and BCL-6. IgH-PCR in this case yielded a single clonal band in the primary tumor. The recurrent tumor showed a novel dominant band in addition to that observed in the primary tumor. Patient 7 presented initially with a DLBCL involving the right humerus. The initial tumor did not appear to express either BCL-2 or BCL-6 but expressed weak p53. The recurrent lymphoma acquired a more aggressive morphology with strong expression of BCL-2 and p53.
Molecular Features
The molecular data are summarized in Table 1, and the results of IgH-PCR analysis are shown in Figure 3. β-globin–positive DNA was available in 18 cases. In two cases (Case 12 and Case 21), no amplifiable DNA was obtained. IgH-PCR demonstrated a dominant band indicative of a clonal B-cell population in 13 of the 18 cases analyzed. All cases were negative for bcl-2/JH gene rearrangement by PCR (data not shown) using MBR and MCR primers. Two of six BCL-6–positive cases were negative for IgH gene rearrangement.

Agarose gel electrophoresis of IgH-PCR. Arrow indicates dominant bands within the 150- to 200-bp range. Cases that were interpreted as negative are listed.
DISCUSSION
PNHLB was first established as a clinical entity when Parker and Jackson described it as “reticulum cell sarcoma” (3). PNHLB is rare and can involve any bony site, but the most common sites of involvement are long bones of the extremities, flat bones of the shoulder and pelvis, and remaining axial skeleton and cranial and jaw bones, in descending order of frequency (4, 9, 10). The male–female ratio ranges from 1.0 to 2.4:1 in many large studies in the United States (1, 5, 25). Male children also appear to be affected more frequently, with a 6:1 ratio. The age range is 1 year and 6 months to 86 years old, with median age range from 36 to 52 years old. The lesions are usually osteolytic and localized but may involve more than one site. Symptoms usually include bone pain and possibly a palpable mass or pathologic fracture, without B symptoms. Estimated long-term survival ranges from 50 to 90%. Although the cases included in this study were from adult patients ranging in age from 25 to 86 years, the male–female ratio, sites of involvement, and presentations are similar to those reported in the literature.
PNHLB have been described as morphologically heterogeneous, and the overwhelming majority of cases have been characterized as B-cell lineage by immunohistochemical techniques (10, 25). Between 69 and 100% of cases have been reported as DLBCL (4, 5, 26) using the REAL classification. Despite numerous reports describing clinicopathologic features of PNHLB, few studies have characterized this group of neoplasms at the immunophenotypic and molecular level. In this study, we analyzed the immunophenotypic and molecular features of 20 cases of PNHLB and compared them with those of DLBCLs occurring in nodal and other extranodal sites. Similar to results of previous studies, there was a preponderance of DLBCLs that was established by morphologic, immunophenotypic, and molecular techniques. Similar to the case of previous reports, a large proportion of our cases exhibited nuclear multilobation (25). No correlation was found between nuclear multilobation and p53, BCL-2, or BCL-6 expression.
To determine possible germinal center derivation of these lymphomas, we performed immunohistochemical studies for BCL-6. The BCL-6 gene encodes a zinc-finger protein that controls germinal center B-cell proliferation and differentiation (27) and is expressed in most normal and neoplastic counterparts of the germinal-center B cell (15, 28, 29, 30, 31). Strong nuclear BCL-6 expression was observed in 6 of 20 cases. The expression of BCL-6 in this subset is supportive of their follicle center origin. CD10 is another antigen that is expressed in neoplastic follicular lymphoma cells. Several reports have demonstrated, however, that only 50% of large B-cell lymphomas, including those arising from bone (32), express CD10 (33, 34), suggesting that it may not be a sensitive marker for cells of germinal center derivation. Another item of potential evidence to support their follicle center origin would be the presence of ongoing somatic mutations of the IgH variable region gene (35). The absence of IgH gene rearrangement in two of the BCL-6–positive cases provides further support for their follicle center origin. However, a detailed molecular analysis of immunoglobulin genes in this group of large-cell lymphomas would provide more information regarding their relationship to the germinal center.
Genetic alterations involving bcl-2 and p53 have been implicated in the pathogenesis of aggressive B cell lymphomas (13), and their expressions serve as important indicators of overall and disease-free survival (36). To determine whether these oncoproteins were involved in the pathogenesis of PLBCL of bone and to compare them with those seen in nodal and other extranodal DLBCLs, we determined the presence of bcl-2/JH gene rearrangements as well as expression of BCL-2 and p53 by immunohistochemistry. Strong BCL-2 expression was seen in 11 of 20 (55%;) cases in the absence of a bcl-2/JH gene rearrangement. The current data support the existence of alternate mechanisms for BCL-2 protein up-regulation in PLBCL of bone, similar to a subset of nodal cases (37). This is supported by other studies that reported that although bcl-2/JH gene rearrangements occur in 17 to 39% of nodal DLBCLs, they are a relatively rare event in extranodal DLBCLs (36). An inverse relationship between BCL-2 and BCL-6 protein expression has been described in the normal secondary follicle (15), whereas in DLBCLs, this relationship is lost (31). Our data showing six cases of PLBCL of bone that demonstrated BCL-2 and BCL-6 co-expression are consistent with that observation and further supports their follicle center derivation.
The p53 tumor suppressor gene product is expressed in 20 to 50% of diffuse, aggressive B-cell lymphomas (13, 14). Its protein expression in lymphomas is associated with histologic transformation (38, 39), treatment failure, and poor outcome irrespective of its mutational status (40). In our study, p53 protein was expressed in the majority (16 of 20; 5 with + expression, 11 with ++ to +++ expression; 80%) of our cases, supporting its role in the pathogenesis of this group of neoplasms. Its role as a prognostic indicator, however, remains to be determined.
An inverse relationship between p53 and BCL-2 protein expression has been described in follicular lymphoma and primary gastric lymphoma (41). Other studies have described co-expression of BCL-2 and p53 in follicular lymphomas, in high-grade lesions, and in cases undergoing transformation more often expressing p53 protein and demonstrating molecular evidence of both bcl-2/JH gene rearrangements and p53 mutation (39). In addition, down-regulation of BCL-2 by mutant p53 has been described in breast cancer (42) and may account for their inverse relationship in follicular lymphomas. In our series, all cases demonstrating BCL-2 expression co-expressed p53. Although the prognostic significance of this observation is unknown, it does highlight the presence of multiple molecular aberrations characteristic of these neoplasms. This is also corroborated by the acquisition of BCL-2 and p53 expression in the recurrent tumor of Case 7 and of BCL-2 and BCL-6 expression in the recurrence of Case 1.
Our study of IgH gene rearrangements and bcl-2/JH rearrangements demonstrates that although the majority (72%) showed IgH gene rearrangements, no case showed a bcl-2/JH rearrangement by PCR. The absence of bcl-2/JH rearrangement in all of the cases is surprising, although others have seen a similarly rare occurrence (4%) of bcl-2/JH translocations in extranodal DLBCLs (36). This may indicate that PLBCL of bone may be genetically distinct from nodal DLBCLs but similar to those arising from other extranodal sites.
Finally, one CD30+ DLBCL exhibited classic anaplastic morphology with a neutrophil-rich background. The absence of ALK-1 expression in this case would support the notion that it represents a morphologic spectrum of DLBCLs (43). No T-cell or null-cell ALCLs were seen in our cases, in contrast to results from a recent study of 13 solitary bone lymphomas (5), of which three (23%) were T-cell or null-cell ALCLs. Our data also support the previous observation that T-cell or null-cell ALCLs are relatively uncommon primary tumors of bone, although CD30+ DLBCLs with anaplastic features can be seen.
In summary, we described 20 PNHLBs, the majority of which are DLBCL by immunophenotype and molecular studies. Molecular analysis of PLBCL of bone, which has been previously unreported, demonstrated IgH chain gene rearrangement and absence of bcl-2/JH gene rearrangement in the majority of cases, suggesting molecular distinction from nodal follicle center–derived neoplasms. Similar to the case with DLBCLs of other extranodal and nodal sites, overexpression of BCL-2, BCL-6, and p53 occur frequently in PLBCL of bone and may have a role in their pathogenesis.
References
Desai S, Jambhekar NA, Soman CS, Advani SH . Primary lymphoma of bone: a clinicopathologic study of 25 cases reported over 10 years. J Surg Oncol 1991; 46: 265–269.
Dubey P, Ha CS, Besa PC, Fuller L, Cabanillas F, Murray J, et al. Localized primary malignant lymphoma of bone. Int J Radiat Oncol Biol Phys 1997; 37: 1087–1093.
Parker F, Jackson H . Primary reticulum cell sarcoma of bone. Surg Gynecol Obstet 1939; 68: 45–53.
Heyning FH, Hogendoorn PC, Kramer MH, Hermans J, Kluin-Nelemans JC, Noordijk EM, et al. Primary non-Hodgkin's lymphoma of bone: a clinicopathological investigation of 60 cases. Leukemia 1999; 13: 2094–2098.
Jones D, Kraus MD, Dorfman DM . Lymphoma presenting as a solitary bone lesion. Am J Clin Pathol 1999; 111: 171–178.
Howat AJ, Thomas HUW, Waters KD, Campbell PE . Malignant lymphoma of bone in children. Cancer 1987; 59: 335–339.
Ueda T, Aozasa K, Ohsawa M, Yoshikawa H, Uchida A, Ono K, et al. Malignant lymphomas of bone in Japan. Cancer 1989; 64: 2387–2392.
Susnerwala SS, Dinshaw KA, Pande SC, Shrivastava SK, Gonsalves MA, Advani SH, et al. Primary lymphoma of bone: experience of 39 cases at the Tata Memorial Hospital, India. J Surg Oncol 1990; 44: 229–233.
Baar J, Burkes RL, Bell R, Blackstein ME, Fernandes B, Langer F . Primary non-Hodgkin's lymphoma of bone: a clinicopathologic study. Cancer 1994; 74: 1194–1199.
Baar J, Burkes RL, Gospodarowicz M . Primary non-Hodgkin's lymphoma of bone. Semin Oncol 1999; 26 (3): 270–275.
Hatori M, Ichinohasama R, Myers J, Schiller A, Kokubun S . Flow cytometrical and genotypic analysis of primary non-Hodgkin's lymphoma of bone. Pathol Res Pract 1997; 193: 557–564.
Antillon F, Behm FG, Raimondi SC, Kaste SC, Sandlund JT, Pappo AS . Pediatric primary diffuse large cell lymphoma of bone with t(3;22)(q27;q11). J Pediatr Hematol Oncol 1998; 20 (6): 552–555.
Kramer MH, Hermans J, Parker J, Krol AD, Kluin-Nelemans JC, Haak HL, et al. Clinical significance of bcl-2 and p53 protein expression in diffuse large B-cell lymphoma: a population based study. J Clin Oncol 1996; 14: 2131–2138.
Martinka M, Comeau T, Foyle A, Anderson D, Greer W . Prognostic significance of t(14;18) and bcl-2 gene expression in follicular small cleaved cell lymphoma and diffuse large cell lymphoma. Clin Invest Med 1997; 20 (6): 364–370.
Raible MD, Hsi ED, Alkan S . Bcl-6 protein expression by follicle center lymphomas. A marker for differentiating follicle center lymphomas from other low-grade lymphoproliferative disorders. Am J Clin Pathol 1999; 112: 101–107.
Chan JKC, Ng C-S, Hui P-K, Leung W-T, Sin V-C, Lam T-K, et al. Anaplastic large cell Ki-1 lymphoma of bone. Cancer 1991; 68: 2188–2191.
Lones MA, Sanger W, Perkins SL, Medeiros LJ . Anaplastic large cell lymphoma arising in bone. Arch Pathol Lab Med 2000; 124: 1339–1343.
Nagasaka T, Nakamura S, Medeiros LJ, Juco J, Lai R . Anaplastic large cell lymphomas presented as bone lesions: a clinicopathologic study of six cases and review of the literature. Mod Pathol 2000; 13: 1143–1149.
Hsu S-M, Raine L, Fanger H . Use of avidin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabelled antibody procedures. J Histochem Cytochem 1981; 29: 557–580.
Harris NL, Jaffe ES, Stein H, Banks PM, Chan JK, Cleary ML, et al. A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood 1994; 84: 1361–1392.
Sambrook J, Fritsch EF, Maniatis T . Molecular cloning. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989.
McCarthy KP, Sloane JP, Wiedemann LM . Rapid method for distinguishing clonal from polyclonal B-cell populations in surgical biopsy specimens. J Clin Pathol 1990; 43: 429–432.
Horsman DE, Gascoyne RD, Coupland RW, Coldman AJ, Sa A . Comparison of cytogenetic analysis, Southern analysis, and polymerase chain reaction for the detection of t(14;18) in follicular lymphoma. Am J Clin Pathol 1995; 103: 472–478.
Stetler-Stevenson M, Lim MS . Use of polymerase chain reaction technique to detect the t(14;18) translocation in lymphoid tissue. Methods Mol Med 1998; 14: 331–339.
Pettit CK, Zukerberg LR, Gray MH, Ferry JA, Rosenberg AE, Harmon DC, et al. Primary lymphoma of bone. A B-cell neoplasm with a high frequency of multilobated cells. Am J Surg Pathol 1990; 14: 329–334.
Korkolopoulou P, Pangalis GA, Patsouris E, Boussiotis VA, Kittas C . B-cell lymphoma of large multilobated type: an immunohistochemical study of 8 cases and review of the literature. Leuk Lymphoma 1994; 13: 151–159.
Ye BH, Rao PH, Chaganti RSK, Dalla-Favera R . Cloning of bcl-6, the locus involved in chromosome translocations affecting band 3q27 in B-cell lymphoma. Cancer Res 1993; 53: 2732–2735.
Flenghi L, Ye BH, Fizzotti M, Bigerna B, Cattoretti G, Venturi S, et al. A specific monoclonal antibody (PG-B6) detects expression of the BCL-6 protein in germinal center B cells. Am J Pathol 1995; 147: 405–411.
Falini B, Bigerna B, Pasqulucci L, Fizzotti M, Martelli MF, Pileri S, et al. Distinctive expression pattern of the BCL-6 protein in nodular lymphocyte predominance Hodgkin's disease. Blood 1996; 87: 465–471.
Falini B, Fizzotti M, Pileri S, Liso A, Pasqualucci L, Flenghi L . BCL-6 protein expression in normal and neoplastic lymphoid tissues. Ann Oncol 1997; 8 Suppl2: 101–104.
Skinnider BF, Horsman DE, Dupuis B, Gascoyne RD . Bcl-6 and Bcl-2 protein expression in diffuse large B-cell lymphoma and follicular lymphoma: correlation with 3q27 and 18q21 chromosomal abnormalities. Hum Pathol 1999; 30 (7): 803–808.
Kramer MH, Hermans E, Wijburg K, Philippo K, Geelen E, van Krieken JH, et al. Clinical relevance of BCL2, BCL6, and MYC rearrangements in diffuse large B-cell lymphoma. Blood 1998; 92: 3152–3162.
Gascoyne RD, Adomat SA, Krajewski S, Connors JM, Reed JC . Prognostic significance of Bcl-2 protein expression and Bcl-2 gene rearrangement in diffuse aggressive non-Hodgkin's lymphoma. Blood 1997; 90: 244–251.
Lo Coco F, Gaidano G, Louie DC, Offit K, Chaganti RSK, Dalla-Favera R . p53 mutations are associated with histologic transformation of follicular lymphoma. Blood 1993; 82: 2289–2295.
Nomdedeu JF, Baiget M, Gaidano G, Estivill C, Lasa A, Rubiol E, et al. p53 mutation in a case of blastic transformation of follicular lymphoma with double bcl-2 rearrangement (MBR and VCR). Leuk Lymphoma 1998; 29: 595–605.
Moller MB, Gerdes AM, Skjodt K, Mortensen LS, Pederssen NT . Disrupted p53 function as predictor of treatment failure and poor prognosis in B- and T-cell non-Hodgkin's lymphoma. Clin Cancer Res 1999; 5: 1085–1091.
Nakamura S, Akazawa K, Kinukawa N, Yao T, Tsuneyoshi M . Inverse correlation between the expression of bcl-2 and p53 proteins in primary gastric lymphoma. Hum Pathol 1996; 27: 225–233.
Haldar S, Negrini M, Monne M, Sabbioni S, Croce CM . Down-regulation of bcl-2 by p53 in breast cancer cells. Cancer Res 1994; 54: 2095–2097.
Haralambieva E, Pulford KA, Lamant L, Pileri S, Roncador G, Gatter K, et al. Anaplastic large-cell lymphomas of B-cell phenotype are anaplastic lymphoma kinase (ALK) negative and belong to the spectrum of diffuse large B-cell lymphomas. Br J Haematol 2000; 109: 584–591.
Moller MB, Gerdes AM, Skjodt K, Mortensen LS, Pederssen NT . Disrupted p53 function as predictor of treatment failure and poor prognosis in B- and T-cell non-Hodgkin's lymphoma. Clin Cancer Res 1999; 5: 1085–1091.
Nakamura S, Akazawa K, Kinukawa N, Yao T, Tsuneyoshi M . Inverse correlation between the expression of bcl-2 and p53 proteins in primary gastric lymphoma. Hum Pathol 1996; 27: 225–233
Haldar S, Negrini M, Monne M, Sabbioni S, CM. C . Down-regulation of bcl-2 by p53 in breast cancer cells. Cancer Res 1994; 54: 2095–2097.
Haralambieva E, Pulford KA, Lamant L, Pileri S, Roncador G, Gatter K, et al. Anaplastic large-cell lymphomas of B-cell phenotype are anaplastic lymphoma kinase (ALK) negative and belong to the spectrum of diffuse large B-cell lymphomas. Br J Haematol 2000; 109: 584–591.
Acknowledgements
Financial support was received from the Sunnybrook and Women's College Health Sciences Center. The authors acknowledge the excellent technical assistance of Eva Peschke and of Donna Ahmad, Dyan Martin, Dyan Cawood, and Marlis McClary of the SCPMG Orange County Medical Library.
Author information
Authors and Affiliations
Corresponding author
Additional information
Megan S. Lim, M.D., Ph.D., is now affiliated with the Department of Pathology, University of Utah, Salt Lake City, Utah.
Rights and permissions
About this article
Cite this article
Huebner-Chan, D., Fernandes, B., Yang, G. et al. An Immunophenotypic and Molecular Study of Primary Large B-Cell Lymphoma of Bone. Mod Pathol 14, 1000–1007 (2001). https://doi.org/10.1038/modpathol.3880425
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880425
Keywords
This article is cited by
-
MR features of primary bone lymphoma in children
Pediatric Radiology (2023)
-
Primary bone lymphoma of the left radius: a case report and related literature review
European Journal of Medical Research (2014)
-
Les lymphomes osseux primitifs au Maroc. Étude rétrospective à propos de 12 cas
Journal Africain du Cancer / African Journal of Cancer (2011)
-
Primary lymphoma of bone: a case report and review of the literature
Medical Oncology (2011)
