
Abstract
Usual interstitial pneumonia is the most common idiopathic chronic interstitial pneumonia, characterized by a temporally heterogenous pattern of interstitial injury with interstitial mononuclear infiltrates, septal fibromyxoid nodules, and parenchymal scarring. This report details the presence of focal eosinophilic pneumonia in six cases of usual interstitial pneumonia in the absence of known causes of this reaction. The relationship of eosinophilic infiltrates in usual interstitial pneumonia with regard to pathogenesis, differential diagnosis, and prognosis is discussed.
Similar content being viewed by others
INTRODUCTION
Usual interstitial pneumonia (UIP) is the most common histologic pattern of chronic interstitial injury seen in patients with clinical idiopathic pulmonary fibrosis (1, 2, 3, 4, 5, 6). Although this pattern of interstitial damage may have a variety of causes, most cases are cryptogenic. UIP has a distinctive histology with a peripheral subpleural and periseptal distribution of patchy interstitial injury that displays temporal heterogeneity—zones of mononuclear cell infiltration alternate with areas of old scarring, with honeycomb fibrosis and fibromyxoid connective tissue nodules within the interstitium. The mononuclear infiltrate consists largely of lymphocytes, especially T cells, and of plasma cells, with frequent lymphoid follicles. In most cases, eosinophils are quite inconspicuous, although they have been noted in up to 25% of patients examined histologically (7). Studies of bronchoalveolar lavage fluid (BALF) in UIP have shown that a level of eosinophils >3% is associated with a poor response to steroids, more severe functional abnormalities, and a worse prognosis (8, 9, 10, 11, 12, 13, 14, 15, 16). Despite the increased percentages of eosinophils in up to 45% of idiopathic UIP cases studied by BALF, the significance of eosinophils in tissue sections has been largely ignored (17, 18, 19, 20). In this report, six patients with idiopathic UIP are described who had patchy zones of eosinophilic pneumonia superimposed on the underlying UIP.
MATERIALS AND METHODS
The consultation files of the author and the Department of Pathology at the University of Pittsburgh Medical Center were searched for all cases of UIP in which prominent eosinophils were noted. Of 124 patients with UIP, 17 cases were identified. In six of these cases, eosinophils consolidated air spaces in a patchy distribution, and these cases form the basis of this report. To be considered eosinophilic pneumonia, it was required that there be (1) more than three sites of discrete alveolar consolidation and (2) filling of >100 air spaces with eosinophils at each site. Eosinophilic pneumonia-like areas occupied approximately 10–30% of tissue available for review in the six cases. Medical records were obtained from patient files and contributing physicians, and follow-up information and drug histories were specifically requested from primary case providers. In no instance was there clinical evidence that the cause of the patient’s pulmonary disease was drug induced, according to the referring pulmonologist or pathologist. Similarly, no patient had a history of connective tissue or autoimmune disease or had documented previous or current parasitic infection.
Records focused on clinical findings at presentation, smoking history, white blood cell counts with cell differential, radiographic abnormalities, therapy, and survival/outcome data. Information was obtained on all six patients. Open-lung biopsies were performed in five patients, and single-lung transplantation with explant examination was done in one patient.
RESULTS
The clinical data on the six patients in the study are presented in Table 1. Five of the six patients were men, with an average age of 49 years (range: 38–58 y). All patients presented with longstanding pulmonary complaints (average: 2.1 y; range: 0.7–4.6 y), chiefly progressive shortness of breath and dyspnea on exertion that had worsened recently. A dry cough accompanied these symptoms in four of the six patients (67%). No patient gave a history of recent infection, although Patient 3 had a history of a pneumonic process treated with trimethoprim-sulfamethoxazole 4 months before biopsy. No evidence of asthma or wheezing was described in the clinical histories. Five patients were cigarette smokers. Laboratory studies showed that no patient (none of six) had a history of peripheral blood eosinophilia, antinuclear antibodies (none of five), anti-neutrophil cytoplasmic antibodies (none of two), or rheumatoid factor (none of three). No patient had a known autoimmune disorder. No patient was on a known pulmonotoxic drug. In four studies, pulmonary function tests were reported to show restrictive lung disease.
Reports of radiologic studies, including chest radiographs and high-resolution computed tomography scans, were available in five cases. All five reports emphasized the bilateral, particularly basilar, reticulonodular infiltrates associated with honeycomb cystic change in the subpleural areas. Three reports described focal ground-glass infiltrates in a patchy random distribution. No emphasis on the peripheral nature of these alveolar infiltrates was made.
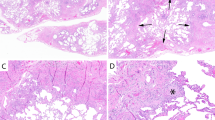
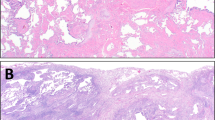
The lung biopsies and explants showed the characteristic features of usual interstitial pneumonia. At low magnification, there was a subpleural and periseptal pattern of parenchymal scarring that culminated in zones of honeycomb fibrosis (Fig. 1). The pulmonary interstitium was expanded by a patchy infiltrate of small round and centrocyte-like lymphocytes associated with plasma cells. In the subpleural zones, the pulmonary lobules were disrupted by broad bands of dense eosinophilic scar tissue accompanied by mononuclear cells and lymphoid aggregates in six of seven (86%) cases. At the advancing front of this dense scar tissue and in zones of mononuclear cell infiltration were foci of young loose fibromyxoid connective tissue containing a proliferation of reactive spindle fibroblasts/myofibroblasts within the edematous matrix (Fig. 2). At the edges of these foci, alveolar pneumocytes were reactive. In these areas of septal injury, air space macrophages were prominent, as they were in the peribronchiolar zones of the cigarette smokers. The temporal heterogeneity of interstitial injury and subpleural and peripheral lobular distribution was typical of UIP. Pertinent negative findings included the absence of granulomas, ferruginous bodies, cannibalistic giant cells, anthracosilicotic nodules, and pleuritis.

Usual interstitial pneumonia. Low magnification demonstrates the patchy, predominantly subpleural distribution of interstitial injury (hematoxylin and eosin staining, 40 ×).

Usual interstitial pneumonia. Zones of damage were characterized by a nonuniform pattern of fibrosis with dense irreversible scarring, interstitial organization (arrows), and mononuclear infiltrates (hematoxylin and eosin staining, 48 ×).
Against this background of UIP were multifocal patchy areas, especially adjacent to subpleural zones of fibrosis, of air space consolidation by bilobed eosinophils (Fig. 3). Macrophages were admixed in all cases and often contained cytoplasmic granules resembling those in the eosinophils. Loose granulation tissue was noted within air spaces in two instances. In these sites of eosinophilic pneumonia, the interstitium contained prominent intravascular and septal eosinophils. No Charcot-Leyden crystalloids, filaria, eggs, fungi, or protozoa were seen with routine or special, such as silver, stains.

Eosinophilic pneumonia. Air space filling by eosinophils with microabscess formation (inset) and macrophages were diagnostic (hematoxylin and eosin staining, 120 ×; inset, 480 ×).
DISCUSSION
Since Liebow’s initial description of the categories of chronic interstitial pneumonias, idiopathic UIP has remained the most common cause of clinical idiopathic pulmonary fibrosis (1, 2). In recent years, emphasis has focused on identifying pathologic mimics of UIP (e.g., bronchiolitis obliterans organizing pneumonia, acute interstitial pneumonia, nonspecific interstitial pneumonia) in an attempt to better define the clinicopathologic significance and behavior of these idiopathic patterns of lung injury (1, 7). With this effort has come only minor changes in our understanding of the histologic spectrum of UIP itself. Particular emphasis has focused on acute exacerbations and organizing pneumonia-like injury patterns in UIP that are accompanied by relatively rapid declines in clinical pulmonary function (21, 22, 23, 24). This report describes one peculiar histologic manifestation of idiopathic UIP with coexistent eosinophilic pneumonia.
The histologic pattern of injury recognized as UIP can be caused by a variety of conditions, and yet there are extremely few histologic features that suggest the underlying etiology. Eosinophilic infiltrates raise the possibility of connective tissue disease and drug-induced lung disease, and yet the presence of eosinophils in idiopathic UIP has been recognized in lavage studies for many years (10, 15, 25). A recent study found rare eosinophils in 25% of cases of UIP (7). In this current report, the six patients with UIP had no clinical evidence of drug injury or autoimmune disease and manifested a distinct consolidation of alveolar spaces and pulmonary parenchyma histologically identical to eosinophilic pneumonia.
The significance of eosinophilic pneumonia in UIP is unclear. From BALF studies, increases in eosinophils are seen in up to 45% of UIP cases and appear to be a marker of more severe impairment of pulmonary function and of poor response to immunosuppressive therapy (10, 11, 12, 13, 14, 17, 26). Histologic correlations with lavage studies fail to show associations of BALF eosinophilia with specific morphologic patterns of injury (8, 14, 18, 19, 20). Unfortunately, none of our six patients had bronchoalveolar lavage performed before biopsy. BALF eosinophilia is often seen with connective tissue disorders, especially scleroderma, acquired immunodeficiency syndrome, mucous hypersecretion, drug-induced lung disease, asthma, and chronic eosinophilic pneumonia (16, 17, 26). These possibilities were excluded in this series.
It is of some interest that four of the six patients in this study were 50 years of age or younger. In studies of UIP, most patients are diagnosed in their mid to late fifties (3). This raises the possibility that some early presentations with UIP may be tied to this unusual variant described in this report.
Recent clinical and radiologic studies of UIP have described markers of activity in UIP that relate to zones of interstitial injury and clinical decompensation (9, 27, 28). Clinically, UIP patients often have an acute onset of fever and dyspnea that evolves over days to weeks and that resembles a persistent viral infection in the terminal stages of their disease. High-resolution computed tomography scans show peripheral zones of ground-glass opacity superimposed on background changes of chronic lung damage, especially honeycomb fibrosis (9, 28). Rare morphologic correlates of this radiographic change have been described, but these areas are thought to show increased interstitial mononuclear cells, air space organization, and hyaline membrane formation (27, 28, 29). Diffuse alveolar damage is observed when extensive portions of both lungs are affected. This study suggests that eosinophilic pneumonia may be one histologic correlate of these zones of activity.
The pathogenesis of idiopathic UIP is unclear but has most often been attributed to mononuclear, especially T-cell, injury to the pulmonary interstitium (30, 31). Activation of T cells is accompanied by elevations of cytokines, including IL-5, that may be chemotactic for other inflammatory cells, including eosinophils (32, 33, 34, 35, 36, 37, 38, 39). Eosinophils elaborate a variety of agents that have a known cytotoxic effect on pulmonary epithelium, including eosinophilic cationic protein and major basic protein, and others that are fibrogenic, especially TGF-β (35, 36, 37).
Treatment of isolated eosinophilic pneumonia requires steroids. Such therapy would seem to be indicated in these special cases as part of conventional therapy for aggressive chronic interstitial pneumonias. There is some data too that cyclophosphamide is helpful in reducing cell counts and eosinophil numbers in the BALF of UIP patients (15).
Eosinophilic pneumonia occurring in the setting of idiopathic UIP raises several differential diagnostic possibilities (40, 41, 42, 43, 44). First, Langerhans’ cells histiocytosis (LCH) is one cause of interstitial scarring that may have a prominent eosinophilic component. The nodular aggregates of Langerhans’ cells in a bronchiolocentric distribution differs from the subpleural, peripheral lobular, and periseptal pattern of interstitial fibrosis in UIP, and Langerhans’ cells do not form large coalescent aggregates in UIP. Pneumothorax can occur in both LCH and idiopathic UIP and may be one cause of pleural and subpleural air space collections of eosinophils (45). Similarly, reactive airway disease inpatients with UIP could have increased peribronchiolar and interstitial eosinophils, but alveolar eosinophils would be unusual. Cigarette smoking has been associated with increased BALF eosinophils in the setting of UIP, but masses of air space eosinophils are not seen in smokers’ bronchiolitis. Chronic eosinophilic pneumonia, if untreated, can lead to irreversible pulmonary fibrosis (43, 46). These patients tend to have a history of asthma and peripheral blood eosinophilia and evanescent episodes of a febrile debilitating illness and would not have the progressive downhill clinical course and unique pattern of subpleural, temporally heterogenous scarring of UIP.
In summary, this report describes six patients with UIP who had coexistent eosinophilic pneumonia as part of their histologic presentation. The interstitial and air space damage associated with air space eosinophils may represent an atypical form of active interstitial injury in some cases of idiopathic UIP.
References
Katzenstein AL, Myers JL . Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med 1998; 157: 1301–1315.
Liebow AA . Definition and classification of interstitial pneumonias in human pathology. Prog Respir Res 1975; 8: 1–31.
Carrington CB, Gaensler EA, Coutu RE, Fitz Gerald MX, Gupta RG . Natural history and treated course of usual and desquamative interstitial pneumonia. N Engl J Med 1978; 298: 801–809.
Crystal RG, Fulmer JD, Roberts WC, Moss ML, Line BR, Reynolds HY . Idiopathic pulmonary fibrosis. Clinical, histologic, radiographic, physiologic, scintigraphic, cytologic, and biochemical aspects. Ann Intern Med 1976; 85: 769–788.
King TE Jr . Idiopathic pulmonary fibrosis. Interstitial lung disease. Am Rev Respir Dis 1993; 2: 367–403.
Liebow AA, Carrington CB . The interstitial pneumonias. In: Simon M, Potchen EJ, LeMay M, editors. Frontiers of pulmonary radiology. New York: Saunders; 1969. p. 102–141.
Travis WD, Matsui K, Moss J, Ferrans VJ . Idiopathic nonspecific interstitial pneumonia: prognostic significance of cellular fibrosing patterns: survival comparison with usual interstitial pneumonia and desquamative interstitial pneumonia. Am J Surg Pathol 2000; 24: 19–33.
Watters LC, Schwarz MI, Cherniack RM, Waldron JA, Dunn TL, Stanford RE, et al. Idiopathic pulmonary fibrosis. Pretreatment bronchoalveolar lavage cellular constituents and their relationships with lung histopathology and clinical response to therapy. Am Rev Respir Dis 1987; 135: 696–704.
Wells AU, Hansell DM, Rubens MB, Cullinan P, Haslam PL, Black CM, et al. Fibrosing alveolitis in systematic sclerosis. Bronchoalveolar lavage findings in relation to computed tomographic appearance. Am J Respir Crit Care Med 1994; 150: 462–468.
Haslam PL, Turton CWG, Lukoszek A, Salsbury AJ, Dewar A, Collins JV, et al. Bronchoalveolar lavage fluid cell counts in cryptogenic fibrosing alveolitis and their relation to therapy. Thorax 1980; 35: 328–339.
Wells AU, Cullinan P, Hansell DM, Rubens MB, Black CM, Newman-Taylor AJ, et al. Fibrosing alveolitis associated with systematic sclerosis has a better prognosis than lone cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med 1994; 149: 1583–1590.
Rudd RM, Haslam PL, Turner-Warwick M . Cryptogenic fibrosing alveolitis. Relationships of pulmonary physiology and bronchoalveolar lavage to response to treatment and prognosis. Am Rev Respir Dis 1981; 124: 1–8.
Peterson MW, Monick M, Hunninghake GW . Prognostic role of eosinophils in pulmonary fibrosis. Chest 1987; 92: 51–56.
Boomars KA, Wagenaar SS, Mulder PGH, van Velzen-Blad H, van den Bosch JM . Relationship between cells obtained by bronchoalveolar lavage and survival in idiopathic pulmonary fibrosis. Thorax 1995; 50: 1087–1092.
Turner-Warwick M, Haslam PL . The value of serial bronchoalveolar lavages in assessing the clinical progress of patients with cryptogenic fibrosing alveolitis. Am Rev Respir Dis 1987; 135: 26–34.
Hiwatari N, Shimura S, Sasaki T, Aikawa T, Ando Y, Ishihara H, et al. Prognosis of idiopathic pulmonary fibrosis in patients with mucous hypersecretion. Am Rev Respir Dis 1991; 143: 182–185.
Allen JN, Davis WB, Pacht ER . Diagnostic significance of increased bronchoalveolar lavage fluid eosinophils. Am Rev Respir Dis 1990; 142: 642–647.
Hyde DM, King TE Jr, McDermott T, Waldron JA Jr, Colby TV, Thurlbeck WM, et al. Idiopathic pulmonary fibrosis. Quantitative assessment of lung pathology. Comparison of a semiquantitative and a morphometric histopathologic scoring system. Am Rev Respir Dis 1992; 146: 1042–1047.
Cherniack RM, Colby TV, Flint A, Thurlbeck WM, Waldron JA Jr, Ackerson L, et al. Correlation of structure and function in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1995; 151: 1180–1188.
Ashcroft T, Simpson JM, Timbrell V . Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol 1988; 41: 467–470.
Panos RJ, Mortenson RL, Niccoli SA, King TE Jr . Clinical deterioration in patients with idiopathic pulmonary fibrosis: causes and assessment. Am J Med 1990; 88: 396–404.
Wells AU, du Bois RM . Prediction of disease progression in idiopathic pulmonary fibrosis. Eur Respir J 1994; 7: 637–639.
Terriff BA, Kwan SY, Chan-Yeung MM, Muller NL . Fibrosing alveolitis: chest radiography and CT as predictors of clinical and functional impairment at follow-up in 26 patients. Radiology 1992; 184: 445–449.
Nagata N, Nagatomo H, Yoshii C, Nikaido Y, Kido M . Features of idiopathic pulmonary fibrosis with organizing pneumonia. Respiration 1997; 64: 331–335.
Schwartz DA, Helmers RA, Dayton CS, Merchant RK, Hunninghake GW . Determinants of bronchoalveolar lavage cellularity in idiopathic pulmonary fibrosis. J Appl Physiol 1991; 71: 1688–1693.
Wells AU, Hansell DM, Haslam PL, Rubens MB, Cailes J, Black CM, et al. Bronchoalveolar lavage cellularity: lone cryptogenic fibrosing alveolitis compared with the fibrosing alveolitis of systemic sclerosis. Am J Respir Crit Care Med 1998; 157: 1474–1482.
Kondoh Y, Taniguchi H, Kawabata Y, Yokot T, Suzuki K, Takagi K . Acute exacerbation in idiopathic pulmonary fibrosis. Analysis of clinical and pathologic findings in three cases. Chest 1993; 103: 1808–1812.
Muller NL, Staples CA, Miller RR, Vedal S, Thurlbeck WM, Ostrow DN . Disease activity in idiopathic pulmonary fibrosis: CT and pathologic correlation. Radiology 1987; 165: 731–734.
Muller NL, Coiby TV . Idiopathic interstitial pneumonias: high-resolution CT and histologic findings. Radiographics 1997; 17: 1016–1022.
McDonald JA . Idiopathic pulmonary fibrosis. A paradigm for lung injury and repair. Chest 1991; 99: 87S–93S.
Crouch E . Pathobiology of pulmonary fibrosis. Am J Physiol 1990; 259: L159–L184.
Hallgren R, Samuelsson T, Venge P, Modig J . Eosinophil activation in the lung is related to lung damage in adult respiratory distress syndrome. Am Rev Respir Dis 1987; 135: 639–642.
Hallgren R, Bjermer L, Lundgren R, Venge P . The eosinophil component of the alveolitis in idiopathic pulmonary fibrosis. Signs of eosinophil activation in the lung are related to impaired lung function. Am Rev Respir Dis 1989; 139: 373–377.
Petrek M, Pantelidis P, Southcott AM, Lympany P, Safranek P, Black CM, et al. The source and role of RANTES in interstitial lung disease. Eur Respir J 1997; 10: 1207–1216.
Noguchi H, Kephart GM, Colby TV, Gleich GJ . Tissue eosinophilia and eosinophil degranulation in syndromes associated with fibrosis. Am J Pathol 1992; 140: 521–528.
Boomars KA, Schweizer RC, Zanen P, van den Bosch JM, Lammers JWJ, Koenderman L . Eosinophil chemotactic activity in bronchoalveolar lavage from idiopathic pulmonary fibrosis is dependent on cytokine priming of eosinophils. Eur Respir J 1998; 11: 1009–1014.
Davis WB, Fells GA, Sun XH, Gadek JE, Venet A, Crystal RG . Eosinophil-mediated injury to lung parenchymal cells and interstitial matrix. A possible role for eosinophils in chronic inflammatory disorders of the lower respiratory tract. J Clin Invest 1984; 74: 269–278.
Fujimoto K, Kubo K, Yamaguchi S, Honda T, Matsuzawa Y . Eosinophil activation in patients with pulmonary fibrosis. Chest 1995; 108: 48–54.
Kroegel C, Foerster M, Grahmann PR, Braun R . Eosinophil priming and migration in idiopathic pulmonary fibrosis. Eur Respir J 1998; 11: 999–1001.
Buchheit J, Eid N, Rodgers G Jr, Feger T, Yakoub O . Acute eosinophilic pneumonia with respiratory failure: a new syndrome. Am Rev Respir Dis 1992; 145: 716–718.
Carrington CB, Addington WW, Goff AM, Madoff IM, Marks A, Schwaber JR, et al. Chronic eosinophilic pneumonia. N Engl J Med 1969; 280: 787–798.
Gaensler EA, Carrington CB . Peripheral opacities in chronic eosinophilic pneumonia: the photographic negative of pulmonary edema. AJR Am J Roentgenol 1977; 128: 1–13.
Jederlinic PJ, Sicilian L, Gaensler EA . Chronic eosinophilic pneumonia. A report of 19 cases and a review of the literature. Medicine 1988; 67: 154–162.
Naughton M, Fahy J, Fitz Gerald MX . Chronic eosinophilic pneumonia. A long-term follow-up of 12 patients. Chest 1993; 103: 162–165.
Luna E, Tomashefski JF Jr, Brown D, Clarke RE, Kleinerman J . Reactive eosinophilic pulmonary vascular infiltration in patients with spontaneous pneumothorax. Am J Surg Pathol 1994; 18: 195–199.
Yoshida K, Shijubo N, Koba H, Mori Y, Satoh M, Morikawa T, Ave S . Chronic eosinophilic pneumonia progressing to lung fibrosis. Eur Respir J 1994; 7: 1541–1544.
Acknowledgements
The author acknowledges the secretarial assistance of Diana Winters and the photographic expertise of Linda Shab.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yousem, S. Eosinophilic Pneumonia-like Areas in Idiopathic Usual Interstitial Pneumonia. Mod Pathol 13, 1280–1284 (2000). https://doi.org/10.1038/modpathol.3880234
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880234
