
Abstract
CD4+ T cells recognizing dietary gluten epitopes in the context of disease-associated human leukocyte antigen (HLA)-DQ2 or HLA-DQ8 molecules are the key players in celiac disease pathogenesis. Here, we conducted a large-scale single-cell paired T-cell receptor (TCR) sequencing study to characterize the TCR repertoire for two homologous immunodominant gluten epitopes, DQ2.5-glia-α2 and DQ2.5-glia-ω2, in blood of celiac disease patients after oral gluten challenge. Despite sequence similarity of the epitopes, the TCR repertoires are unique but shared several overall features. We demonstrate that clonally expanded T cells dominate the T-cell responses to both epitopes. Moreover, we find V-gene bias of TRAV26, TRAV4, and TRBV7 in DQ2.5-glia-α2 reactive TCRs, while DQ2.5-glia-ω2 TCRs displayed significant bias toward TRAV4 and TRBV4. The knowledge that antigen-specific TCR repertoire in chronic inflammatory diseases tends to be dominated by a few expanded clones that use the same TCR V-gene segments across patients is important information for HLA-associated diseases where the antigen is unknown.
Similar content being viewed by others


Introduction
Celiac disease is a chronic inflammatory disease of the small intestine caused by hypersensitivity to dietary gluten in genetically disposed individuals who are human leukocyte antigen (HLA)-DQ2 (95%) or HLA-DQ8.1 Activation of CD4+ T cells that recognize gluten peptides presented on HLA-DQ2 or HLA-DQ8 is an important step in the celiac disease pathogenesis. The intestinal inflammation is characterized by lymphocyte infiltration, villus atrophy, and crypt hypertrophy.2 Patients in active disease have circulating IgA and/or IgG antibodies to both gluten and the autoantigen transglutaminase 2.3 Maintaining a gluten-free diet will normalize the intestinal architecture and the antibody titers in most cases. However, a small but detectable number of gluten-reactive memory CD4+ T cells are present in the blood of treated celiac disease patients even after years of adherence to a gluten-free diet.4 Upon re-introduction of dietary gluten for 3 days in the gluten-challenge scheme, there is a concerted activation of memory gluten-reactive CD4+ T cells. On day six after gluten challenge, a large efflux of recently activated gluten-reactive CD4+ T cells is detectable in blood by enzyme-linked immunoSpot (ELISPOT) assays or by tetramer staining.5, 6 Moreover, most of these gluten-reactive T cells have been shown to express gut-homing β7 integrin5 and the activation marker CD38.7
There are several recent reports on the T-cell receptor (TCR) usage as well as TCR:pMHC crystal structures of gluten-reactive CD4+ T cells from celiac disease patients.8, 9, 10 Among the HLA-DQ2-restricted gluten-reactive T cells, the TCRs reactive to DQ2.5-glia-α2 are best characterized,11, 12 and crystal structures of three DQ2.5-glia-α2-reactive TCRs were recently published.9 Crystal structures give atomic insight into recognition of a few pMHC complexes. This insight is complemented by TCR sequencing of whole populations of T cells specific for the pMHC complexes with solved structures as well as of T cells specific for structurally related pMHC complexes. For this reason we have undertaken a large-scale global repertoire analysis of DQ2.5-glia-α2-reactive TCRs, and contrasted this with analysis of the TCR repertoire of T cells specific for the homologous gluten epitope DQ2.5-glia-ω2. We performed paired TCR sequencing of tetramer-sorted CD4+ T cell reactive to these two immunodominant epitopes directly ex vivo from three gluten-challenged celiac disease patients. The DQ2.5-glia-α2 and DQ2.5-glia-ω2 epitopes have six identical amino acid residues in the 9-mer core region, but differ in the P5 (L vs. Q) and P7 (Y vs. F) positions that were shown to be important TCR contact points in the published crystal structures. Our data showed that the DQ2.5-glia-α2 and DQ2.5-glia-ω2 TCR repertoires were unique and non-overlapping but shared overall features such as clonal dominance and biased usage of some TRAV and TRBV gene segments that were shared across individuals. The knowledge that antigen-specific TCR repertoire in celiac disease tends to be dominated by a few expanded clones expressing same TCR V-gene segments across patients is vital information for other HLA-associated diseases where the antigen is unknown.
Results
Validation of tetramer sorting and TCR sequencing
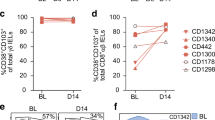
Peripheral blood mononuclear cells from three treated celiac disease patients after gluten challenge were stained with HLA-DQ2.5-tetramers presenting the DQ2.5-glia-α2 and DQ2.5-glia-ω2 epitopes (Table 1) and tetramer-binding CD4+ effector-memory T cells were sorted (Supplementary Figure S1online) for in vitro expansion or for ex vivo single-cell analysis. The frequencies of DQ2.5-glia-α2 and DQ2.5-glia-ω2 tetramer-positive effector-memory CD4+ T cells after gluten challenge were found to range between 48 and 654, and 105–255 per million CD4+ T cells, respectively (Figure 1). The antigen specificity of tetramer sorting was validated in a subset of sorted cells that were cultured in vitro as T-cell clones (TCCs) by antigen-free expansion and cloning by limited dilution.4 On re-testing of their antigen specificity, 91% (132/145) of TCCs retained the specific tetramer-staining and 67% (100/149) retained specific proliferative response in peptide-stimulation assays. This data shows that the T cells we sorted with tetramers were specific.

Tetramer staining of peripheral blood CD4+ T cells. Peripheral blood mononuclear cells (PBMCs) from three celiac disease patients were stained with DQ2.5-tetramer representing the DQ2.5-glia-α2 and DQ2.5-glia-ω2 epitopes, followed by bead enrichment of the tetramer-positive cells. Single DQ2.5-glia-α2- or DQ2.5-glia-ω2- tetramer-binding CD4+ effector-memory T cells were sorted. (a) Tetramer staining of CD4+ T cells with DQ2.5-tetramers representing the DQ2.5-glia-α2 and DQ2.5-glia-ω2 epitopes in all three patients. (b) Frequency of tetramer-positive effector-memory CD4+ T cells per million total CD4+ T cells.
In the TCR data generated from direct ex vivo sequencing of single tetramer-sorted T cells, we observed dual V-gene usage in 3.7% of the unique clones analyzed. We have not observed wells in which there was both dual T-cell receptor alpha (TRA) and dual T-cell receptor beta (TRB) sequences suggesting that sorting of two T cells in the same well was a rare phenomenon. Identical triplets of dual TRA sequences together with one unique TRB sequence was often observed in different wells containing single-cells sorted from the same individual, showing that the observed dual V-gene usage was not a technical artefact. Moreover, the single-cell TCR sequencing data is validated by multiple examples of identical pairs of TRA and TRB sequences obtained by Sanger sequencing of in vitro generated TCCs from T cells sorted from the same individual.
Clonal dominance in DQ2.5-glia-α2- and DQ2.5- glia-ω2-reactive CD4+ T cells
Paired TCR sequences were grouped in clonotypes and in both DQ2.5-glia-α2- and DQ2.5-glia-ω2-reactive T cells of all three patients, top three unique clones accounted for roughly half of the TCR repertoire (Figure 2). In both repertoires, the most dominant clones showed V-gene bias shared between patients. Unique clone expressing TRBV7-2 paired with TRAV26-1 or TRAV9 was observed as one of the three most dominant clones across all the patients in DQ2.5-glia-α2-reactive T cells. Similarly, TRAV12 and TRAV1 were preferentially expressed in the dominant clones across DQ2.5-glia-ω2-reactive T cells in two of three patients.

Clonal dominance in DQ2.5-glia-α2- and DQ2.5-glia-ω2-reactive CD4+ T cells. Each slice represents a unique clone and the number of T cells belonging to that unique clone is shown in the slices. The slices without number represent unique clones that were observed only once.
TCRs of both DQ2.5-glia-α2- and DQ2.5-glia-ω2-reactive T cells exhibit biased V-gene usage
For TRAV- or TRBV- usage analysis, we analyzed both paired and unpaired TRA or TRB sequences. In the case of dual V-gene usage, the V gene with highest number of clones (if obtained from cloning) or with highest number of reads (if obtained from high-throughput sequencing) was used for analysis. In some cases of dual V-gene usage in clonally expanded population, we carried out functional analysis of hybridoma cells with retrovirally transduced TCR to identify the V gene involved in antigen recognition and used the functional V gene for analysis (Supplementary Figure S2).
In each individual patient, the three most frequently used TRAV and TRBV genes accounted for around half of the unique clones in both DQ2.5-glia-α2- and DQ2.5-glia-ω2-TCR repertoires indicating presence of biased V-gene usage (Supplementary Figure S3). Despite inter-individual variation on the dominantly used V-gene segments, same TRAV and TRBV gene segments were preferentially expressed across the patients.
TRA sequencing of 227 DQ2.5-glia-α2-reactive T cells from three patients produced altogether 59 unique clones expressing 18 different TRAV genes (Table 2). The two most frequently used TRAV genes were TRAV26 (29%) and TRAV4 (15%; Figure 3a). Comparison of TRAV usage with a reference library of single tetramer-negative CD4+ T cells from HLA-DQ2.5+ individuals (344 TCRα and 388 TCRβ sequences) showed that this bias was significant (TRAV26 (P=0.0001) and TRAV4 (P=0.0005)). Similar analysis of DQ2.5-glia-ω2-reactive T cells showed that 25 different TRAV genes were used (Table 2). The two most frequently used TRAV genes in DQ2.5-glia-ω2 TCR repertoire were TRAV12 (24%) and TRAV4 (14%; Figure 3b). However, this bias was only significant for TRAV4 (P=0.0005) but not for TRAV12 (P=0.12).

Usage of V-gene segment in DQ2.5-glia-α2 (a,c) and DQ2.5-glia-ω2 (b,d) TCR repertoire. To analyze the TRAV (a,b) and TRBV (c,d) usage, each unique clone obtained from all the patients using the same TRAV or TRBV genes were grouped in one category. Each slice represents a unique TRAV or TRBV gene. The percentage of unique clones using them is shown in the slice for genes that were used above 5%. Diverse TRAVs or TRBVs expressed only once are represented by the slices without any color and number.
Analysis of TRB sequences of 234 DQ2.5-glia-α2-reactive T cells from three patients generated 63 unique clones expressing 14 different V genes (Table 2). The two most frequently used TRBV genes in DQ2.5-glia-α2 TCR repertoire were TRBV7 (51%, P=0.0001) and TRBV20 (11%, not significant; Figure 3c). In comparison, the two most frequently used TRBV genes (of total 18 different TRBV genes we obtained) in DQ2.5-glia-ω2 TCR repertoire were TRBV4 (24%, P=0.0001) and TRBV7 (13%, not significant; Figure 3d).
The TRBV7 bias in DQ2.5-glia-α2-TCR repertoire was contributed mainly by TRBV7-2 in contrast to that in the DQ2.5-glia-ω2-TCR repertoire. Although 41% of the unique clones in DQ2.5-glia-α2-TCR repertoire express TRBV7-2, it is only 5% in the DQ2.5-glia-ω2-TCR repertoire. This observation is in agreement with the estimates for the frequency of TRBV7-2 obtained by staining with a Vβ6.7-specific antibody where we observed that 19–29% of DQ2.5-glia-α2-reactive T cells were Vβ6.7-positive, compared with 0.65–14% of DQ2.5-glia-ω2-reactive T cells (data not shown).
In brief, V-gene bias shared between patients was observed in both repertoires suggesting that this phenomenon could be a hallmark of gluten epitope-reactive TCR repertoire.
Preferential TRAV/TRBV pairing is only observed in DQ2.5-glia-α2 reactive T cells
Unique TRAV/TRBV pairs were analyzed to confirm previously reported preferential TRAV26-1/TRBV7-2 pairing in the DQ2.5-glia-α2-reactive repertoire, and to investigate pairing preference among DQ2.5-glia-ω2-reactive T cells (Table 2).
Around half of total unique TCCs across all the patients expressed the three most frequently used unique TRAV/TRBV pairs in the DQ2.5-glia-α2-TCR repertoire (Figure 4a). We confirm the previous observation of preferential TRAV26-1/TRBV7-2 pairing. In addition, TRAV4/TRBV7-2 preference was also observed in the DQ2.5-glia-α2-TCRs. This is an interesting observation since TRAV26-1 and TRAV4 are phylogenetically two closely related V genes with 70% sequence identity.

TRAV/TRBV pairing in DQ2.5-glia-α2 (a) and DQ2.5-glia-ω2 (b) TCR repertoire. Each slice of the bar represents a unique TRAV/TRBV pair and the number of unique clones expressing them is shown in slices. Diverse TRAV/TRBV pairs expressed only once are represented by the slices without any color or number.
In contrast, TCRs of DQ2.5-glia-ω2-reactive T cells did not exhibit any striking bias in TRAV/TRBV pairing (Figure 4b). The 16 (CD578), 25 (CD737), and 15 (CD774) unique TCCs analyzed were found to use 16, 21, and 14 unique TRAV/TRBV pairs, respectively, exhibiting no preferential pairing. In addition, we did not find any TRAV26-1/TRBV7-2 TCRs in the DQ2.5-glia-ω2 repertoires.
Alanine substitution analysis
As the TCR repertoires of DQ2-glia-α2- and DQ2.5-glia-ω2-reactive T cells despite great epitope homology appeared to be differently composed, we wanted to understand if there is a difference of epitope recognition by the DQ2.5-glia-α2 and DQ2.5-glia-ω2-reactive T cells. To this end, proliferative responses of TCCs toward single alanine-substituted variants of their wild-type epitope were measured. The TCR sequences of the TCCs used are given in Table 3.
For the alanine scan analysis, we selected 10 DQ2.5-glia-α2-reactive TCCs that were only reactive toward DQ2.5-glia-α2 and not to other closely related epitopes including DQ2.5-glia-ω2 (data not shown). Although the effect of alanine substitutions was TCC dependent, we found that alanine substitution of the P7 tyrosine completely abolished the response of all DQ2.5-glia-α2-reactive TCCs, in accordance with the previous observation.9 In addition, alanine substitution of P5 leucine and P3 proline largely abrogated the response in the majority of TCCs. Although P1 and P8 substitutions also reduced the reactivity of the majority of the clones, the effect of P4 and P6 substitutions was more diverse. Furthermore, the alanine substitutions at P9, P10, and P11 had almost no effect on the reactivity of the clones (Figure 5a and Supplementary Figure S4A).

Reactivity patterns of all DQ2.5-glia-α2-reactive (a) and DQ2.5-glia-ω2-reactive TCCs (b) to the single alanine-substituted peptides normalized to the response to the wild-type peptides. All measurements were performed in triplicate. Error bars represent mean±s.d.
The effect of alanine substitution was relatively similar across the 8 DQ2.5-glia-ω2-reactive TCCs analyzed (Figure 5b and Supplementary Figure S4B). All these clones were reactive toward DQ2.5-glia-ω2 and not to DQ2.5-glia-α2. Alanine substitution at P3 proline, P5 glutamine and P7 phenylalanine largely abolished the responses of all DQ2.5-glia-ω2-reactive TCCs. Loss of reactivity caused by P4 glutamate substitution was observed in all the clones but was not as strong as observed for the previous three positions. In general, substitution at positions P2 through P8 resulted in reduction or loss of reactivity in the majority of the clones. Furthermore, substitutions at positions P-1, P1, P10, and P11 were dispensable for TCR recognition.
In a nutshell, while P7 tyrosine is critical for DQ2.5-glia-α2 recognition, P3 proline, P5 glutamine, and P7 phenylalanine are critical for DQ2.5-glia-ω2 recognition.
Scrutiny of cross-reactivity
A small population of cross-reactive T cells that bind both DQ2.5-glia-α2 and DQ2.5-glia-ω2 tetramers was found in all three patients. T cells binding both tetramers were sorted from patients CD578 and CD774 and cultured in vitro. Upon re-staining, TCCs cultured from the cross-reactive populations from both patients were tetramer-binding. However, only TCCs from CD774 showed proliferative response to both DQ2.5-glia-α2 and DQ2.5-glia-ω2 peptides, whereas TCCs from CD578 were only reactive to DQ2.5-glia-α2 but not to DQ2.5-glia-ω2 in T-cell proliferation assays. In patient CD737, although cross-reactive cells were not sorted as a separate population, we found that some of the cells sorted as DQ2.5-glia-ω2-tetramer-positive cells were cross-reactive in T-cell assays after in vitro culture and expansion. In all three patients, the striking feature of these cross-reactive populations was the extreme degree of clonal expansion where we found one clone in two patients and two clones in the third patient. It is noteworthy that the dominant clone in two of three patients used TRAV4 (Figure 6a).

Reactivity pattern of cross-reactive TCCs. TCR sequences of the clonally expanded cross-reactive T cells used for analysis are shown in a. The reactivity pattern of cross-reactive T cells to the single alanine-substituted peptides normalized to the response to the wild-type peptides are summarized in b,c. Reactivity of TCCs against chimeric peptides: α2_L5Q, ω2_Q5L, α2_Y7F, and ω2_F7Y is shown in d. Responses were normalized to the response of wild-type peptides. Response of cross-reactive clones against peptides representing the epitopes DQ2.5-glia-α2 (α2), DQ2.5-glia-ω2 (ω2), DQ2.5-glia-α1 (α1; QLQPFPQPELPY, underlined 9-mer core amino acid sequence), DQ2.5-sec2 (sec2; GQQPFPQPEQPFPQSG), DQ2.5-glia-ω1 (ω1; PQQPFPQPEQPFP), DQ2.5-γ3 (γ3; FPQQPEQPYPQQP), DQ2.5-glia-γ4a (γ4a; FSQPEQEFPQPQ), and DQ2.5-glia-γ4b (γ4b; FPQPEQEFPQPQ) (e). All measurements were performed in triplicate. Error bars represent mean±s.d.
To understand how these cross-reactive clones interact with the two epitopes, proliferative responses of the clonally dominant cross-reactive clones from CD737 (TCC737.A4B30) and CD774 (TCC774.A4B9) were tested in alanine scan (Figure 6a). We found that the cross-reactive clones interacted with the DQ2.5-glia-α2 or DQ2.5-glia-ω2 epitopes with the same pattern as the non-cross-reactive TCCs. P7 tyrosine was most important for the recognition of DQ2.5-glia-α2, followed by P3 proline, while the effect of P5 leucine substitution varied (Figure 6b and Supplementary Figure S4C). Similarly, in case of DQ2.5-glia-ω2, loss of reactivity with P7 phenylalanine substitution and overall loss or reduction of reactivity due to substitution at positions P2 through P8 was observed (Figure 6c and Supplementary Figure S4D).
To further elucidate what makes these TCCs cross-reactive, we analyzed the proliferation in response to single amino acid-exchanged chimeric peptides. As P9 was dispensable for reactivity in both the epitopes, we focused on the P5 and P7 positions. We generated four chimeric peptides: α2_L5Q, ω2_Q5L, α2_Y7F, and ω2_F7Y. The cross-reactive TCCs showed reactivity toward all the chimeric variants of peptides at various intensities (Figure 6d and Supplementary Figure S4C,D). In contrast, the epitope-specific TCCs did not recognize the chimeric peptides (Figure 6d and Supplementary Figure S4A,B). This indicates that the ability of the cross-reactive clones to recognize both leucine and glutamine at P5, and both tyrosine and phenylalanine at P7, unlike the epitope-specific TCCs, allow them to be cross-reactive.
Moreover, proliferative response of the cross-reactive clones toward other closely related gluten epitopes was investigated. We found that in addition to DQ2.5-glia-α2 and DQ2.5-glia-ω2, the cross-reactive clones were strongly reactive to the DQ2.5-sec2 peptide that contained a single tryptophan-to-glutamine substitution at P9 of the core sequence compared with DQ2.5-glia-ω2. TCC774.A4B9 was also reactive to DQ2.5-γ3, which is an epitope that is very similar to the α2_L5Q chimeric peptide. The responses toward other epitopes (DQ2.5-glia-α1, DQ2.5-glia-ω1, DQ2.5-γ4a, and DQ2.5-γ4b) were none or very low (Figure 6e). This observation suggests that they are not promiscuous, but specifically reactive to DQ2.5-glia-α2 and DQ2.5-glia-ω2 and other highly similar peptide variants.
Discussion
This study by single-cell TCR sequencing reports that the effector-memory T-cell response to two immunodominant gluten T-cell epitopes in celiac disease, DQ2.5-glia-α2 and DQ2.5-glia-ω2 exhibit clonal dominance and biased usage of some TRAV and TRBV gene segments that are shared between different individuals.
We sorted tetramer-binding antigen-specific T cells in celiac disease patients 6 days after a 3-day gluten-challenge regime. Among the tetramer-positive cells we found evidence of clonal expansion as for each of the epitopes three unique clones made up roughly half of the TCR repertoire. Some degree of clonal expansion had most likely taken place during the 3-day gluten antigen exposure in these patients who otherwise were maintaining a gluten-free diet. However, as all three patients participating in this study were diagnosed as adults, we believe that much of the clonal expansion was already present in the memory T-cell pool resulting from gluten exposure before the diagnosis. A recent study of children with celiac disease reported that TCRs against DQ2.5-glia-α1/α2 epitopes also exhibit clonal expansion of a few dominant clones13 similarly to what is observed in adult CD patients. Our preliminary results moreover indicate that clonally dominant TCRs of peripheral blood are found also in biopsies of treated CD patients after gluten challenge as well as in biopsies of untreated CD patients (unpublished data; Risnes LF, Dahal-Koirala S, Sarna VK, Lundin KEA, Sollid LM and Qiao SW). Thus taken together, these observations suggest that a few clonally expanded clones dominate the T-cell response to the gluten and these dominant clones populate both the intestinal tissue and blood. There is reason to believe that clonal dominance of the antigen-specific T-cell response is most likely a phenomenon common to many chronic inflammatory conditions. This notion is supported by sequencing studies performed on populations of T cells reactive to complex antigens such as viruses14 and bacteria,15, 16 or to unknown antigens in autoimmune conditions17, 18, 19 or single-cell sequencing of antigen-specific T cells that express one biased used TRBV gene.20 In the current study, we have directly shown clonal dominance to one single pMHC complex by unbiased sequencing of the TCR repertoire on the single cells without in vitro expansion. To our knowledge, such studies of antigen-specific CD4+ T cells involved in human diseases are scarce.
Another striking feature of the TCR repertoires we analyzed was the prevalence of biased usage of certain TRAV and TRBV genes that are shared across individuals. In the DQ2.5-glia-α2-reactive TCR repertoire, we replicated previous findings of TRBV7-2 and TRAV26-1 bias, with the addition of TRAV4 that interestingly is phylogenetically closely related to TRAV26. TRAV4 and TRAV26-1 share the expression of Tyr38α, which in all the three published ternary structures of TRAV26-1/TRBV7-2 TCR in complex with HLA-DQ2.5:DQ2.5-glia-α2 makes contacts with several residues of the β chain of HLA-DQ2.5 (ref. 9). In the DQ2.5-glia-ω2-reactive TCR repertoire, we found statistically significant biased usage of TRAV4 and TRBV4. Although the TRAV12-3 gene segment was frequently used, it was not statistically significant when compared with the usage of the TRAV12 gene family, in particular TRAV12-2, in our reference TCR repertoire generated from tetramer-negative CD4+ T cells from HLA-DQ2.5+ individuals. The biased usage of TRAV4 that was observed in both TCR repertoires and the prevalent usage of TRAV12 in CD4+ T cells in general, indicate that the common restriction element, namely HLA-DQ2.5, may impose some of the TCR repertoire bias. Larger data sets of TCR repertoires reactive to different HLA-DQ2.5-restricted epitopes will be needed to clarify the HLA-DQ2.5 signature on the TCR repertoire.
Crystal structures of TCR: HLA-DQ2.5:DQ2.5-glia-α2 complexes show that the TCRs interact with P5 leucine and P7 tyrosine of the DQ2.5-glia-α2 that are two of the three residues that differ between DQ2.5-glia-α2 and DQ2.5-glia-ω2.9 In light of this, it is not surprising that we found different TCR repertoires reactive to DQ2.5-glia-α2 and DQ2.5-glia-ω2. Similarly, it also sheds light on the observation that only cross-reactive TCCs recognize amino acid variants of both the epitopes in P5 and P7 positions.
In the published ternary structures of TRAV26-1/TRBV7-2 TCR in complex with HLA-DQ2.5:DQ2.5-glia-α2, the germline-encoded regions of the TRBV7-2-encoded TCRβ chain showed limited interaction with the pMHC.9 Likewise, single-site mutations of germline-encoded residues of the TCRβ chain had modest effects on binding to HLA-DQ2.5:DQ2.5-glia-α2. Thus, the molecular reason for the strong bias for TRBV7-2 of TCRs that recognize DQ2.5-glia-α2 remains elusive. In line with published sequencing data, we found an Arg residue in the CDR3β of 92% of the TRBV7-2-encoded DQ2.5-glia-α2-reactive TCRs. Crystal structures have shown that this non-germline-encoded Arg109β residue acts as the lynchpin that mediates recognition of DQ2.5-glia-α2. However, we did not find an increased usage of Arg, or any other conserved sequence motifs, in the CDR3α of TRAV12- or TRAV4- encoded TCRαs, or in the CDR3β of TRBV4-encoded TCRβs that were involved in the recognition of DQ2.5-glia-α2 or DQ2.5-glia-ω2, respectively.
Single-cell analysis enabled us to study the pairing between TCRα and TCRβ chains. In the DQ2.5-glia-α2-reactive repertoire, we found that the frequently used TRAV26-1 and TRBV7-2 genes were also preferentially used as a pair, in agreement with published data.9, 11, 12 In the DQ2.5-glia-ω2 TCR repertoire, however, we were not able to detect any preferential pairing. This is puzzling as we on the other hand found frequent use of TRAV12 (24%), TRAV4 (14%), and TRBV4 (24%) in this TCR repertoire. It might be that these V genes are frequently used because they are capable of making functional pair with many partner V genes and thus do not show pair preferences. We suspect that there may be less dominant pairing preferences that will only become apparent in even larger data sets of paired TCR sequences.
In conclusion, despite differences in the DQ2.5-glia-α2- and DQ2.5-glia-ω2-TCR repertoires, they show both biased responses shared among patients and high degree of clonal expansion. We hypothesize that clonally expanded TCRs that use the same TCR V-gene segments across patients may be a common feature for most antigen-reactive TCR repertoires that are involved in chronic diseases. Studying TCRs that carry these characteristics can be a fruitful strategy for identifying yet unknown causative antigens in chronic inflammatory diseases with strong HLA associations.
Methods
Patients
Oral gluten challenge was carried out in three celiac disease patients (CD578, CD737, and CD774). The patients were on a gluten-free diet when given a 3-day gluten challenge before a blood sample was taken 6 days later.12 The patients have given informed written consent and the study was approved by the regional ethics committee (S-06114a).
Tetramer staining, cell enrichment, and FACS
Tetramerized recombinant HLA-DQ2.5 covalently linked with gluten-derived peptides containing the T-cell epitopes DQ2.5-glia-α2 (PQPELPYPQPE, underlined 9-mer core amino acid sequence) and DQ2.5-glia-ω2 (FPQPEQPFPWQP; Table 1) were multimerized on phycoerythrin-labeled streptavidin (Invitrogen, Waltham, MA) or allophycocyanin-labeled streptavidin (ProZyme, Hayward, CA).
The tetramer-binding CD4+ T cells were bead-enriched from peripheral blood followed by sorting on FACS Aria II (Becton Dickinson, Franklin Lakes, NJ) according to the protocol described in Christophersen et al.4 Cells within the singlet lymphocyte gate were further scrutinized to identify the tetramer-binding cells that were: CD3+, CD11c-, CD14-, CD19-, CD56-, CD45RA-/CD62L-, and CD4+. As a result, tetramer-binding CD4+ effector-memory T cells were sorted for in vitro expansion or for ex vivo single-cell analysis (Supplementary Figure S1).
T-cell proliferation assay
Based on previously established protocol, the sorted cells were cloned by limited dilution and expanded without antigens.4 The specificity of the growing TCCs was tested by re-staining with gliadin tetramers and analyzed on a FACS Calibur (BD Biosciences), and in T-cell proliferation assay.
In T-cell proliferation assays, Epstein–Barr virus-immortalized B-lymphoblastoid cell line from an HLA-DR3, DQ2 homozygous celiac disease patient (CD114) was used as allophycocyanins. In brief, 75,000 allophycocyanins were irradiated 75 Gy and incubated with 10 μM antigen peptides at 37 °C for 24 h before the addition of 50,000 T cells. After another 48 h, cultures were pulsed with 1 μCi 3H-thymidine and harvested 16–20 h later. Stimulation index was calculated by dividing counts per minute after antigen stimulation with counts per minute after medium stimulation. TCCs with stimulation index value above three for a peptide were identified to be peptide specific. All the peptides used had purity >70%.
TCR sequencing
Two different approaches based on template switch were used to sequence paired TRA and TRB genes.
TCR sequencing from in vitro cultured TCCs
Based on previously published Switching mechanism at the 5′-terminus of the RNA transcript (SMART) protocol, total RNA from TCCs cultured from tetramer-sorted T cells were used in complementary DNA (cDNA) synthesis together with a template-switch oligo.21 Total RNA was isolated with RNeasy Mini kit (Qiagen, Hilden, Germany). Total RNA of 40–100 ng was reverse transcribed with 1.2 μM of reverese transcription oligo (d(AAGCAGTGGT ATCAACGCAGAGTAC)-r(GGG)), 2 mM of dithiothreitol, 1 mM of deoxynucleotide (dNTP), 1.2 μM of oligo dT (16; 5′-CTGAATTCTTTTTTTTTTTTTTTT-3′), RNAsin and 10 U μl–1SuperScript II (Invitrogen) in a total reaction volume of 10 μl. The cDNA synthesis was done in two steps, where in the first step total RNA and oligo dT(16) in a total reaction of 5 μl was subjected to 72 °C for 3 min and 42 °C for 2 min to allow breaking of double strands. In the second step, all the other reagents were added and cDNA was synthesized at 42 °C for 90 min with a final inactivation at 72 °C for 10 min. The PCR was performed with 1 μl of cDNA, 200 μM of dNTP, 500 nM of SMART forward primer (5′-AAGCAGTGGTATCAACGCAGAGT-3′), 500 nM of reverse primer (TRAC_rev (5′-GAACTTTCTGGGCTGGGGAAGAAGGTGTCTTCTGG-3′), or TRBC_rev (5′-TGC TTCTGATGGCTCAAACACAGCGACCT-3′)) and Phusion polymerase (Thermo Scientific, Waltham, MA) in total 25 ul, for 5 cycles (98 °C for 10 s and 72 °C for 60 s), then 5 cycles (98 °C for 10 s, 70 °C for 30 s, and 72 °C for 40 s), followed by 22 cycles (98 °C for 10 s, 68 °C for 30 s, and 72 °C for 40 s) and a final elongation at 72 °C for 4 min. The PCR products were then subjected to Sanger sequencing on an ABI3730 machine. The sequences were analyzed with the IMGT/V-QUEST online resource.22 It is a simple and fast approach but sometimes the V gene can be assigned with high degree of certainty, but the crucial CDR3 sequences cannot be defined due to low-sequencing quality.
In those cases where the V-gene usage could be resolved by SMART PCR but the crucial CDR3 sequences could not be defined, we have taken the same template-switch cDNA but used gene-specific forward primers specific to the V gene for the particular chain. This approach gives highly specific products with good-sequencing traces. However, this method required an initial screening with SMART PCR. To this end, two sets of 28 and 18 partially degenerate forward primers (Supplementary Table S1) that anneal to each of the 45 and 48 productive human TRAV and TRBV genes were prepared. The PCR was done with 1 μl of cDNA, 200 μM of dNTP, 500 nM of gene-specific TRAV, or TRBV forward primers, 500 nM of reverse primer (TRAC_rev*(5′-AGTCAGATTTG TTGCTCCAGGCC-3′) or TRBC_rev* (5′-TTCACCCACCAGCTCAGCTCC-3′)) and Phusion polymerase in total 25 ul, for 8 cycles (98 °C for 20 s, 60 °C for 40 s, and 72 °C for 40 s), followed by 22 cycles (98 °C for 20 s, 55 °C for 40 s, and 72 °C for 40 s) and a final elongation at 72 °C for 3 min. PCR products were sequenced on an ABI3730 machine and the sequences were analyzed with the IMGT/V-QUEST online resource.
In cases where we could not resolve V-gene usage by SMART PCR or in the cases of dual V-gene expression, we have amplified the PCR product a few more cycles to incorporate enzyme digestion sites and subsequently cloned them into a cloning vector. Miniprep DNA was then sequenced an ABI3730 machine. This sequencing method gives good quality sequencing traces and is helpful in identifying dual V-gene usage. However, this method is labor-intensive.
TCR sequencing from ex vivo-sorted single cells
To sequence TCR genes from sorted single cells, we used a modified “template switch” protocol based on two published studies.21, 23 In short, we first prepared full-length cDNA from FACS-sorted single cells followed by semi-nested PCR to obtain gene-specific PCR products, i.e., TRA and TRB sequences. Each plate has 96-well-specific barcodes and plate-specific barcodes for both TRA and TRB sequences, located at the 5′end and 3′ end, respectively.
A 96-well cell capture plate (Bio-Rad, Hardshell PCR plates, Hercules, CA), containing 5 μl of buffer (100 mM Tris-HCl at pH 8, 375 mM KCl, 30 mM MgCl2, 0.1% Tween-20) with 400–800 nM Bio-STRT-T30VN rev oligo (5′-Bio-TTAAGCAGTGGTATCAACGCAGAGTCGACTTTTTTTTTTTTTTTTTTTTTTTTTTTTTVN-3′) and 400–700 nM STRT-n (Barcoded template-switch oligo; 5′-AAGCAG TGGTATCAACGCAGAGTGCAGUGCUNNNNNNrGrGrG-3′, where “rG” denotes a riboguanine and “NNNNNN” a well-specific 6-bp barcode) was prepared according to Islam et al.20 Single CD4+ T cells were sorted in 96-well capture plates. Each plate was immediately frozen on dry ice to facilitate cell lysis. First-strand cDNA synthesis followed by cDNA purification step with magnetic beads was carried out according to Islam et al.20 The purified full-length cDNA pooled from all 96 wells of a plate, was then amplified in a two-step process following the same protocol.
Two rounds of semi-nested PCR, with separate reaction mix for TRA and TRB was carried out. The reaction mix for the first PCR (2 μl of full-length cDNA obtained after 10-cycle amplification, 200 μM dNTP, 200 nM forward primer (STRT-fwd (5′-CGAGATAAGCAGTGGTATCAACGCAGAGTGC-3′), 200 nM reverse primer (TRAC_rev or TRBC_rev) with Advantage 2 Polymerase mix in a total volume of 25 μl) was amplified for 1 min × 95 °C followed by 16 cycles (15 s × 95 °C, 30 s × 65 °C, and 1 min × 68 °C) with final elongation at 68 °C for 3 min. The second PCR was done in a total volume of 25 μl with 2 μl of the first PCR product, 200 μM dNTP, 200 nM forward primer (STRT-fwd), 200 nM reverse primer (STRT-TRAC_ADP2 (5′-AGTTCAGACGTGTGCTCTTCCGATCT XXXXXXCAGCTGGTACACGGCAGGG-3′), or STRT-TRBC_ADP2 (5′-AGTTCAGACGTGTGC TCTTCCGATCTXXXXXXCGACCTCGGGTGGGAACAC-3′; where “XXXXXX” stands for plate-specific 6-bp barcode)) and Advantage 2 Polymerase mix for 1 min × 95 °C followed by 15 cycles (15 s × 95 °C, 30 s × 65 °C, and 1 min × 68 °C) with final elongation at 68 °C for 3 min. The set of 10 STRT-TRAC_ADP2 and 10 STRT-TRBC_ADP2 reverse primers used is listed in Supplementary Table S2. A 5-μl aliquot was amplified for another 10 cycles and visualized on a 1.5% agarose gel to confirm the size of the PCR product (∼600 bp).
A final PCR was carried to prepare the library for high-throughput sequencing. PCR was done in a total volume of 25 μl with 2 μl of second PCR product, 200 μM dNTP, 200 nM forward primer (STRT-fwd), 200 nM reverse primer (STRT-seq2 (5′-GACTGGAGTTCA GACGTGTGCTCTTCCGATCT-3′)) and Phusion DNA polymerase for 1 min × 98 °C followed by 12 cycles (15 s × 98 °C, 30 s × 65 °C, and 1 min × 72 °C) with final elongation at 72 °C for 5 min. A 5-μl aliquot was visualized on a 1.5% agarose gel. The intensity of band was analyzed to calculate the quantity of PCR product from each plate, and approximately equal quantity of PCR products from each of the 20 plates (10 for TRA and 10 for TRB) were pooled together. The PCR products were cleaned with QIAquick PCR purification kit (Qiagen), and the PCR library was subjected to high-throughput sequencing (PacBio). High-throughput sequencing data were analyzed in collaboration with the Bioinformatics Core Facility for University of Oslo and Oslo University Hospital. The raw sequence reads were subjected to a sequential process of validation before being analyzed. The sequences were aligned, collapsed, and binned according to the well and plate barcodes. The identity threshold of 0.96 was used to generate consensus sequences of reads with identical plate and well barcode to compensate for PCR error as well as the relatively high-sequencing error rate by PacBio. Only sequences with >5 reads were included in the analysis using IMGT/HighV-QUEST. TRAV and TRBV gene usage was compared using Fisher’s exact test (GraphPad).
References
Sollid, L.M. & Jabri, B. Triggers and drivers of autoimmunity: lessons from coeliac disease. Nat. Rev. Immunol. 13, 294–302 (2013).
Marsh, M.N. Studies of intestinal lymphoid tissue. XI—The immunopathology of cell-mediated reactions in gluten sensitivity and other enteropathies. Scanning Microsc. 2, 1663–1684 (1988).
du Pre, M.F. & Sollid, L.M. T-cell and B-cell immunity in celiac disease. Best Pract. Res. Clin. Gastroenterol. 29, 413–423 (2015).
Christophersen, A. et al. Tetramer-visualized gluten-specific CD4+ T cells in blood as a potential diagnostic marker for coeliac disease without oral gluten challenge. United European Gastroenterol. J. 2, 268–278 (2014).
Raki, M. et al. Tetramer visualization of gut-homing gluten-specific T cells in the peripheral blood of celiac disease patients. Proc. Natl. Acad. Sci. USA 104, 2831–2836 (2007).
Anderson, R.P., Degano, P., Godkin, A.J., Jewell, D.P. & Hill, A.V. In vivo antigen challenge in celiac disease identifies a single transglutaminase-modified peptide as the dominant A-gliadin T-cell epitope. Nat. Med. 6, 337–342 (2000).
du Pre, M.F. et al. CD62L(neg)CD38(+) expression on circulating CD4(+) T cells identifies mucosally differentiated cells in protein fed mice and in human celiac disease patients and controls. Am. J. Gastroenterol. 106, 1147–1159 (2011).
Broughton, S.E. et al. Biased T cell receptor usage directed against human leukocyte antigen DQ8-restricted gliadin peptides is associated with celiac disease. Immunity 37, 611–621 (2012).
Petersen, J. et al. T-cell receptor recognition of HLA-DQ2-gliadin complexes associated with celiac disease. Nat. Struct. Mol. Biol. 21, 480–488 (2014).
Petersen, J. et al. Determinants of gliadin-specific T cell selection in celiac disease. J. Immunol. 194, 6112–6122 (2015).
Qiao, S.W., Christophersen, A., Lundin, K.E. & Sollid, L.M. Biased usage and preferred pairing of a- and ß-chains of TCRs specific for an immunodominant gluten epitope in coeliac disease. Int. Immunol. 26, 13–19 (2014).
Qiao, S.W. et al. Posttranslational modification of gluten shapes TCR usage in celiac disease. J. Immunol. 187, 3064–3071 (2011).
Hardy, M.Y. et al. Consistency in polyclonal T-cell responses to gluten between children and adults with celiac disease. Gastroenterology 149, 1541–1552 (2015).
Bitmansour, A.D. et al. Clonotypic structure of the human CD4+ memory T cell response to cytomegalovirus. J. Immunol. 167, 1151–1163 (2001).
Becattini, S. et al. T cell immunity. Functional heterogeneity of human memory CD4(+) T cell clones primed by pathogens or vaccines. Science 347, 400–406 (2015).
Du, G. et al. TCR repertoire, clonal dominance, and pulmonary trafficking of mycobacterium-specific CD4+ and CD8+ T effector cells in immunity against tuberculosis. J. Immunol. 185, 3940–3947 (2010).
Lu, S. et al. Aneurysmal lesions of patients with abdominal aortic aneurysm contain clonally expanded T cells. J. Immunol. 192, 4897–4912 (2014).
Sakkas, L.I. et al. Oligoclonal T cell expansion in the skin of patients with systemic sclerosis. J. Immunol. 168, 3649–3659 (2002).
Toivonen, R., Arstila, T.P. & Hanninen, A. Islet-associated T-cell receptor-beta CDR sequence repertoire in prediabetic NOD mice reveals antigen-driven T-cell expansion and shared usage of VbetaJbeta TCR chains. Mol. Immunol. 64, 127–135 (2015).
Bowerman, N.A. et al. Identification of multiple public TCR repertoires in chronic beryllium disease. J. Immunol. 192, 4571–4580 (2014).
Quigley, M.F., Almeida, J.R., Price, D.A. & Douek, D.C. Unbiased molecular analysis of T cell receptor expression using template-switch anchored RT-PCR. Curr. Protoc. Immunol. 94 Vii:10.33:10.33.1 -10.33.16 (2011).
Brochet, X., Lefranc, M.P. & Giudicelli, V. IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res. 36, W503–W508 (2008).
Islam, S. et al. Highly multiplexed and strand-specific single-cell RNA 5′ end sequencing. Nat. Protoc. 7, 813–828 (2012).
Acknowledgements
We thank all the patients in the study, Marie K Johannesen for technical assistance and Bjørg Simonsen for tetramer production. This work was supported by grants from the Research Council of Norway through its Centre of Excellence funding scheme, project number 179573/V40 and the South-Eastern Norway Regional Health Authority.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
SUPPLEMENTARY MATERIAL is linked to the online version of the paper .
Supplementary information
Rights and permissions
About this article

Cite this article
Dahal-Koirala, S., Risnes, L., Christophersen, A. et al. TCR sequencing of single cells reactive to DQ2.5-glia-α2 and DQ2.5-glia-ω2 reveals clonal expansion and epitope-specific V-gene usage. Mucosal Immunol 9, 587–596 (2016). https://doi.org/10.1038/mi.2015.147
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2015.147
This article is cited by
-
The methylome of the celiac intestinal epithelium harbours genotype-independent alterations in the HLA region
Scientific Reports (2019)
-
Transglutaminase 2 and Transglutaminase 2 Autoantibodies in Celiac Disease: a Review
Clinical Reviews in Allergy & Immunology (2019)
-
Deep sequencing of blood and gut T-cell receptor β-chains reveals gluten-induced immune signatures in celiac disease
Scientific Reports (2017)
-
T Cell Receptor Profiling in Type 1 Diabetes
Current Diabetes Reports (2017)
-
Coeliac disease: a unique model for investigating broken tolerance in autoimmunity
Clinical & Translational Immunology (2016)
