
Abstract
Investigations of ultrafast processes occurring on the nanoscale require a combination of femtosecond pulses and nanometer spatial resolution. However, controlling femtosecond pulses with nanometer accuracy is very challenging, as the limitations imposed both by dispersive optics on the time duration of a pulse and by the spatial diffraction limit on the focusing of light must be overcome simultaneously. In this paper, we provide a universal method that allows full femtosecond pulse control in subdiffraction-limited areas. We achieve this aim by exploiting the intrinsic coherence of the second harmonic emission from a single nonlinear nanoparticle of deep subwavelength dimensions. The method is proven to be highly sensitive, easy to use, quick, robust and versatile. This approach allows measurements of minimal phase distortions and the delivery of tunable higher harmonic light in a nanometric volume. Moreover, the method is shown to be compatible with a wide range of particle sizes, shapes and materials, allowing easy optimization for any given sample. This method will facilitate the investigation of light–matter interactions on the femtosecond–nanometer level in various areas of scientific study.
Similar content being viewed by others


Introduction
The ability to control ultrafast light pulses with femtosecond and nanometer precision is of crucial importance for the study of fundamental dynamic processes in individual nano-objects, including coherent dynamics in molecules and photosynthetic complexes,1,2 femtosecond exciton dephasing in quantum dots3 and ultrafast responses in plasmonic nanostructures.4,5,6,7
However, there are two fundamental limitations on the confinement of light in the femtosecond and nanometer regimes: on the one hand, femtosecond pulses are prone to becoming distorted in the phase domain while propagating through dispersive elements, such as lenses or objectives. Such distortions typically result in a stretched pulse: i.e., longer than the Fourier limit.8 On the other hand, diffraction prevents light from being focused to sizes smaller than a few hundred nanometers. Controlling light with femtosecond and nanometer precision thus requires Fourier-limited pulses in a subdiffraction limited area.
Owing to its intrinsic coherence, the second harmonic (SH) emitted by a nonlinear crystal is sensitive to the spectral phase of a laser pulse and has therefore been widely used for femtosecond pulse characterization, mainly in combination with frequency-resolved optical gating9 or spectral phase interferometry for direct electric-field reconstruction of ultrashort optical pulses.10 The SH has also been used for both pulse characterization and compression with the so-called multiphoton intrapulse interference phase scan (MIIPS) technique.11,12
In this work, we have used MIIPS as a method for measuring and compensating for the phase distortions of a laser pulse after passage through a high numerical aperture (NA) microscope objective. In its standard implementation, MIIPS makes use of a bulk SH crystal and a pulse shaper to retrieve the spectral phase of a laser pulse and to correct for the phase distortions introduced by the optics. MIIPS is an excellent tool for addressing the femtosecond portion of the problem, and efficient pulse compression has been demonstrated in the focus of a high-NA objective for ultrafast microscopy applications.12 However, even under optimal conditions, the spatial resolution is limited by the size of the focused beam at the sample position, that is, the diffraction limit. As a result, spectral and phase inhomogeneities in the focal volume cannot be addressed by standard MIIPS, which limits the validity of pulse shaping and calibration for ultrafast nanoscale experiments on single molecules, quantum dots, plasmonic systems, nanodiamonds, etc.
To overcome the diffraction limitation, phase information must be obtained based on a signal originating from a truly nanometric volume. To this end, we have used single nonlinear nanoparticles (NPs), instead of bulk crystals, as nanometer sources of SH generation. Because NPs are smaller than the excitation wavelength, SH phase matching conditions do not apply.13 As a consequence, efficient SH generation can be achieved from a very broad wavelength range without the need to specifically tune the NP size or to change their orientation with respect to the incident light. These advantages make NPs more cost effective and easier to handle than their bulk crystal counterpart.
The SH emission from single nonlinear NPs13,14,15,16,17,18 and nanotips19 interacting with ultrafast pulses has recently been detected and studied. Using frequency-resolved optical gating, Extremann et al.14 demonstrated ∼70-fs pulse characterization on single SH NPs. However, frequency-resolved optical gating only allows precise measurements of the spectral phase of a laser pulse; it does not provide active control over the time duration or phase properties of the pulse, which is required for controlled femtosecond–nanometer investigations. Similarly, Wnuk et al.16 presented SH autocorrelation traces from single NPs and studied the SH response for chirped 200-fs and compressed 13-fs pulses, but did not take advantage of the NPs for full femtosecond pulse control. Interestingly, Berweger et al.19 showed full optical waveform control at the apex of a metallic tip by exploiting the concept of adiabatic plasmonic focusing. This approach is promising for the extension of scanning probe microscopy20 to the ultrafast regime; however, it does not provide a general method for pulse calibration and control in lens-based optical microscopes.
To our knowledge, no general calibration method has yet been developed that allows full control of a femtosecond laser pulse over a subdiffraction-limited area. In this paper, we fill this gap and provide such a universal experimental method. Using the SH emitted by a single nonlinear NP, in combination with a pulse shaper, we demonstrate that a femtosecond laser pulse can be efficiently compressed and that its spectral phase can be controlled with high precision using the signal originating from a nanometric volume. Our method is versatile and compatible with a wide variety of microscope systems. Moreover, several advantages are gained by using NPs rather than, e.g., scanning tips as nanometer-sized probes: embedded in any biological or engineered sample, NPs have the ability to function as labels for nano-imaging,21 as probes for local phase effects and short and long-lived quantum coherence and as nanoscale sources of tunable higher harmonic light.
However, for all of these applications, it must first be established that the NP can sustain excitation with the shortest possible pulses without distorting the pulse shape, that minimal phase effects can be measured and that an arbitrary phase shape can reliably be imposed upon a femtosecond pulse in the nanometric volume of the particle. In this paper, we achieve these goals in two steps: first, we demonstrate the compression of a laser pulse down to the Fourier limit (17 fs) using the SH emitted by a single nonlinear NP as small as 150 nm. Second, once the pulse has been compressed, we also show deterministic coherent control of the SH spectrum of a single NP, achieved by accurately shaping the excitation pulse in the phase domain. The robustness and reproducibility of the method are finally demonstrated for NPs of different sizes, shapes and materials.
We consider that our findings will enhance the accuracy and user-friendliness of, e.g., ultrafast microscopy and nanoscopy, multiphoton microscopy and spatially resolved spectroscopy.
Materials and methods
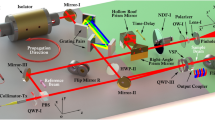
The experimental set-up used herein consists of a broadband titanium sapphire laser (Octavius 85M; Menlo Systems GmbH, Am Klopferspitz 19a D-82152 Martinsried, Germany) tuned to a central wavelength of ∼800 nm, with a bandwidth of ∼90 nm calculated from the points at which the intensity of the spectrum drops to 30% of its maximum value. The repetition rate of the laser is 85 MHz, and the average power at the sample position was varied from 0.5 to 6 mW. The laser was sent through a pulse shaper (adapted from MIIPS-box; Biophotonics Solutions, Inc. Laboratories, 1401 East Lansing Drive, Suite 112 East Lansing, MI 48823, USA) based on a liquid-crystal spatial light modulator (SLM) integrated in a 4f-configuration and into an inverted confocal microscope. The laser beam was focused to a diffraction-limited spot with a 1.3-NA objective (Fluar; Zeiss, Carl-Zeiss Strasse 22 73447 Oberkochen, Germany) onto a thin (150 µm) microscope cover slip containing the SH NPs. A nanometer-precision piezoelectric scanner (Mad City Labs Inc., 2524 Todd Drive Madison, WI 53713, USA) allowed the sample area to be scanned over several microns. The SH from the NPs was collected in reflection through the same 1.3-NA objective and was sent either to an Avalanche photo-diode (Perkin-Elmer, 940 Winter Street Waltham Massachusetts 02451, USA) for imaging purposes or to a spectrograph (Shamrock SR-303i; Andor Technology plc. 7 Millennium Way Springvale Business Park, Belfast BT12 7AL, UK) for spectral measurements, using an electron-multiplying charge-coupled device camera (iXon; Andor). In all of the experiments described, we used linearly polarized excitation, and we collected the total SH signal emitted, integrated over all polarization directions.
We studied three different types of SH NPs: barium titanate (BaTiO3), iron iodate (Fe(IO3)3) and gold (Au) NPs, as illustrated by the scanning electron microscopy (SEM) images in Figure 1. BaTiO3 (Sigma-Aldrich, Laborchemikalien GmbH P.O. Box 100262 30918 Seelze, Germany) and Fe(IO3)3 (PlasmaChem GmbH, Rudower Chaussee 29 D-12489 Berlin, Germany) were dispersed in ethanol, sonicated and deposited onto a glass microscope cover slip and dried. The Fe(IO3)3 NPs have mean dimensions of ∼150 nm and were homogeneously distributed over the coverslip, whereas in the case of BaTiO3, an inhomogeneous distribution of single NPs and small aggregates with sizes ranging from one hundred to a few hundred nanometers was found. Thus, the BaTiO3 sample is suitable for size- and shape-dependent experiments.

SEM images of typical NPs used in this study. (a) BaTiO3; (b) Fe(IO3)3; (c) Au. The scale bar is the same in all images. NP, nanoparticle; SEM, scanning electron microscopy.
Spherical Au NPs, 100 nm in diameter, suspended in water solution and citrate-coated, were purchased from BB International (BBI SOLUTIONS 73 Ty Glas Avenue Cardiff, CF14 5DX, UK) and were prepared by interchanging the coating from citrate to BSPP following the method described by Bidault and Polman.22 Briefly, 50 mL of citrate-coated particles was incubated overnight with 10 mg BSPP. The particles were then centrifuged to remove the supernatant before being rinsed and finally resuspended in a 0.5 mM BSPP solution. The final Au NP concentration was on the order of 0.02 µM. The Au dimer shown in the bottom right image of Figure 1 was formed by aggregation without any linker, and therefore, the two Au NPs were in direct contact.
The experiment was performed as follows: first, optical images of the NPs were acquired by scanning the sample with the piezoelectric scanner and collecting the SH signal with the avalanche photo-diode. Next, while continuously exciting one NP and recording its SH spectrum, we performed MIIPS to measure the spectral phase and to compress the initially distorted pulse down to its Fourier limit.
During a MIIPS iteration, a set of reference phase functions is applied to the SLM and the full SH spectrum is detected, which allows the initial spectral phase to be retrieved. In a second step, the negative of the retrieved phase (known as a compensation mask) is applied to the SLM to correct for distortions, and a new MIIPS iteration is run to measure the new spectral phase and to create a new compensation mask. This process is repeated until a compensation mask that yields a constant spectral phase over the full laser spectrum (i.e., producing a Fourier-limited pulse) is found. For details on the mechanism of MIIPS, see Refs. 11,12 and the references therein.
All of the theoretical SH spectra reported in this paper were simulated using the femtoPulse Master V1.1 software from BioPhotonic Solutions, Inc.
SEM was used to characterize the size and shape of the NPs studied in the optical measurements.
Results and discussion
We first studied the SH response of BaTiO3 NPs, which were taken as our reference sample (Figures 2 and 3). We then tested our method using Fe(IO3)3 and Au NPs to verify the robustness and versatility of the method (Figure 4).

Femtosecond pulse compression on a single 180-nm BaTiO3 NP. (a) SEM image of the investigated area of a BaTiO3 sample. (b) SH image of the sample area before pulse compression. The circled particle was chosen for pulse compression. (c) SH image of the sample area after pulse compression, measured under identical acquisition settings. Note the total SH intensity increase collected from the NPs (the same color bar applies to both SH images). Red arrows indicate small NPs (mean dimension of 125 nm) that are clearly detectable only after pulse compression. (d) Laser spectrum used in the experiment, together with the spectral phase measured before and after pulse compression. Note that MIIPS successfully compensated for ∼40-rad distortions. (e) SH spectra of the selected NP, as measured for different compensation masks during MIIPS optimization. Note that the SH spectrum after compression agrees very well with the simulated result (dashed black line). (f) Simulated (red dashed line) and measured interferometric autocorrelation (black line) of the compressed 17-fs pulse, as explained in the text. Inset: calculated time profile of the uncompressed (dashed red line) and compressed pulse (solid blue line). MIIPS, multiphoton intrapulse interference phase scan; NP, nanoparticle; SEM, scanning electron microscopy; SH, second harmonic.

(a) Laser spectrum and spectral phases measured for different BaTiO3 NPs. (b) Mean spectral phase over 15 measurements. The error bars are given by the standard deviation for the 15 measurements. Note that the total variation of the spectral phase is less than 0.3 rad, and the standard deviation is approximately 0.15 rad. (c) SEM image of the sample, showing the 15 BaTiO3 NPs used for measurement. (d) SH control for NP 8. The SH spectra measured before and after compression and for a step function of amplitude π applied to the SLM are compared with the calculated spectra. MIIPS, multiphoton intrapulse interference phase scan; NP, nanoparticle; SEM, scanning electron microscopy; SH, second harmonic; SLM, spatial light modulator.

Top: SH control on a Fe(IO3)3 NP (a), on a Au dimer (b) and on a single Au NP (c). For each NP, the SH spectra were collected for compressed (black) and uncompressed pulses (green) and for a step function of amplitude π (red) applied to the SLM. These results are shown together with the corresponding simulated SH spectra (dashed lines). For the single Au NP, the wavelength range was increased up to 480 nm to include the TPPL emission. Bottom: spectral phase measured for one (d) Fe(IO3)3 NP (red line) and (e) Au dimer (red line) and Au monomer (blue line) when applying a compensation mask obtained for a BaTiO3 NP. The error bars are the standard deviations obtained from repeated measurements of BaTiO3 NPs. MIIPS, multiphoton intrapulse interference phase scan; NP, nanoparticle; SH, second harmonic; SLM, spatial light modulator; TPPL, two-photon photoluminescence.
Figure 2 shows the results of the pulse characterization and compression using the SH signal from a single BaTiO3 NP. The upper panel shows an SEM image (Figure 2a) and optical images of the same sample area, as measured before (Figure 2b) and after (Figure 2c) pulse compression under otherwise identical experimental conditions. The SH emission from individual NPs is clearly resolved in the optical images. An increase of more than one order of magnitude in the total SH signal from the NPs is observed upon excitation with compressed pulses. The NP highlighted with a circle in Figure 2, which was found to have a mean dimension of 180 nm by SEM, was selected for performing pulse compression.
In Figure 2d, the fundamental laser spectrum used in the experiments is plotted together with the spectral phase retrieved by MIIPS before and after pulse compression. The initial spectral phase was measured to span approximately 40 rad across the entire laser spectrum, reflecting the amount of phase distortion introduced by the optics used in the experiment. After a few MIIPS iterations, a flat spectral phase was obtained with variations of less than 0.3 rad (Figures 2d and 3b), corresponding to nearly Fourier-limited pulses.
Figure 2e shows SH spectra measured for the selected NP after successive MIIPS iterations (i.e., with successive compensation masks applied to the SLM). Each MIIPS iteration is associated with an overall SH intensity increase. The initial low-intensity SH spectrum (red dashed line) is a direct consequence of the high phase distortions initially present in the laser pulse and can intuitively be understood as follows: high phase distortions correspond to a stretched pulse in the time domain, where different frequency components of the laser spectrum arrive at the sample at different times. Because the sum frequency generation of different frequency components cannot occur if the components are not reaching the sample at the same time, the contribution of this nonlinear effect to the SH signal is smaller for a stretched pulse. As MIIPS proceeds and the pulse is re-compressed in time, the SH intensity increases and the spectrum approaches the simulated result corresponding to Fourier-limited pulses (black dashed line). The total SH intensity collected from the NP was found to increase by a factor of 15 between the uncompressed and the Fourier-limited pulse (blue line). This increase is of fundamental importance for detecting even smaller particles, as shown in Figure 2c. The two NPs indicated with red arrows, not detectable using uncompressed pulses, become visible upon excitation with a Fourier-limited pulse. As shown by the SEM images in the right panel of Figure 1a, these particles have mean dimensions of 125 nm.
These results clearly demonstrate our ability to compress femtosecond pulses using the SH produced by a subdiffraction-limited NP. More importantly, the results show that femtosecond pulses can be delivered to nanometric probes unperturbed by, e.g., spatiotemporal coupling23,24 or by the probe itself. Moreover, nonlinear signals and phase information can be reliably extracted from deep subwavelength volumes, which is an important prerequisite for accurate nonlinear nanoscopy.
It is noteworthy that the intensity of the SH signal collected from a single BaTiO3 NP in our experiment was sufficient to produce a SH spectrum in less than 1 s, which allows MIIPS to proceed rapidly. Typically, fewer than 10 MIIPS iterations are needed to compensate for the phase distortions, when using 32 points (i.e., acquiring 32 different SH spectra) for the first four iterations and 128 points for the remaining iterations. Full pulse compression therefore takes less then 15 min.
As an independent verification of the Fourier-limited character of the compressed pulse, we acquired an interferometric autocorrelation trace of the laser pulse using the SH emitted by the same NP. Such an interferometric autocorrelation trace is measured by scanning the relative time delay of two identical replicas of the original pulse generated by the pulse shaper, as described by Pestov et al.25 The graph in Figure 2f shows the integrated SH intensity as a function of the time delay between the pulse replicas. The interferometric autocorrelation function has a full-width at half-maximum (FWHM) of 23.6 fs, which, for a Gaussian pulse, corresponds to a pulse duration of 16.7 fs (FWHM). To illustrate the dramatic effect of the compression on the time profile of the laser pulse, we also plotted the calculated intensity autocorrelation25 for the pulse before and after compression as an inset. For the compressed pulse, a single narrow peak with a FWHM of 23.6 fs is observed, whereas in the case of the uncompressed pulse, a much broader central peak (56-fs FWHM) is observed, superimposed on a very broad pedestal containing 85% of the total integrated intensity and extending up to ±1 ps from the central peak.
To test the reproducibility of this method and its independence of the specific particle, we measured the spectral phase for several NPs differing in size, shape and even material. First, we studied the SH emitted by the 15 different BaTiO3 NPs indicated in the SEM image of Figure 3. These NPs provide a large distribution of sizes, varying from 130 to 500 nm in mean dimension, with a variety of shapes (see representative examples in Figure 1a).
In Figure 3a, we show the spectral phases measured for the 15 different BaTiO3 NPs when applying the best compensation mask previously obtained (namely, the mask corresponding to Fourier-limited pulses) using NP 1 as the SH source. Very good agreement among all of the measured spectral phases was found. If one considers only the area of the fundamental spectrum in which the intensity is above 30% of the maximum, i.e., where the measurement is the most reliable, then the mean phase varies by less than 0.3 rad and the standard deviation is below 0.15 rad (Figure 3b), thus confirming the good level of compensation obtained and the independence of our method with respect to the size and shape of the NP.
The fact that the same phase response was measured for NPs that differ in size, shape and orientation allows us to draw direct conclusions regarding the possible polarization dependence of the spectral phase response and potential spatiotemporal coupling. The polarization of the excitation light is generally used to study the spatial orientation of SH particles (see, e.g., Ref. 13). Here, we used linear polarization to excite many randomly oriented NPs. The process of measuring differently oriented NPs with respect to the polarization axis is equivalent to an effective rotation of the incident polarization. Because the measured spectral phase is consistent among the NPs, we conclude that, in the range of particle sizes explored, our phase measurement is independent of the relative polarization of the incoming light. In addition, the SH NPs, which are smaller than the diffraction-limited excitation spot, provide a means to investigate possible spatiotemporal coupling inside the focal volume. Each NP, depending on its relative position with respect to the focus and its specific size, shape and orientation, probes a different subvolume of the excitation spot. Therefore, the fact that the phase response does not depend on the specific NP investigated ensures that different subvolumes in the excitation spot have the same spectral phase. In other words, there is no measurable spectral phase inhomogeneity inside the focal volume.23,24 The ability to test for spatiotemporal inhomogeneities inside the excitation spot is a unique advantage of using NPs as opposed to SH bulk crystals and provides a tool for correctly designing pulse-shaping experiments on the nanoscale.
In a typical pulse-shaping experiment,1 once a pulse has been compressed, the SLM is used to control the pulses via the application of specific spectral phases, e.g., to obtain chirped pulses or a sequence of pulses with defined delays and phase relations. To demonstrate full pulse control on the nanoscale level, we applied different phase masks to the SLM, acquired corresponding SH spectra and compared them with simulations. The test phases included the flat phase (corresponding to Fourier-limited pulses), the phase before pulse compression (corresponding to distorted pulses) and a mask with a π phase difference between the short and long wavelength parts of the laser spectrum. As Figure 3d confirms, the SH spectra collected for NP 8 in the SEM images (mean size of 160 nm) corresponding to such applied phases are in very good agreement with the calculated spectra. These results also show that the SH emission from a single NP can be deterministically manipulated in its frequency domain. In practice, once our compression method is applied, these NPs can be considered as nanometer sources of easily tunable, coherent blue light, with pulse lengths and wavelengths all controlled by the application of the proper spectral phase.
As a final test of the universal nature of our method, we performed similar measurements on particles composed of Fe(IO3)3 and Au. The results for these NPs are reported in Figure 4. Although pulse compression can also be performed on these particles, starting from a completely uncompressed pulse and obtaining similar results as for the BaTiO3 NPs, the main objective of the experiments described herein is to verify that the same spectral phase is measured regardless of the SH NP material. For this purpose, we proceeded as follows: first, we compressed the laser pulse inside the focus of the microscope objective using BaTiO3 NPs, retrieving a phase with variations of less than 0.3 rad over the laser spectrum, as described above. Then, a few phase measurements were performed on different BaTiO3 NPs to obtain the mean phase and standard deviation. Next, we changed to either the Fe(IO3)3 or the Au sample and ran a phase measurement on one NP, starting from the compensation mask obtained from MIIPS on the BaTiO3 NPs. In Figure 4, these phase measurements together with the SH spectra for different applied phases are shown for an Fe(IO3)3 NP 110 by 160 nm in size (see Figure 1b bottom) and for two different Au NPs: a dimer composed of two attached 100-nm Au spheres and a single 100-nm Au sphere (Figure 1c). For these particles, the laser power was reduced from 6 mW to approximately 0.5 mW to avoid photo damage. Even when applying this lower power to Fe(IO3)3, we were able to collect SH spectra with less than a 1-s integration time for 150-nm particles, confirming the good SH efficiency of this material.14 For the Au NPs, an integration time of less than 1 s was still possible in the case of the Au dimer, whereas for the single 100-nm Au NPs, the integration time was increased to 2 s or longer to obtain an adequate signal-to-noise ratio in the SH spectra.
As illustrated in Figure 4d, for the Fe(IO3)3 NPs, the measured phase is well within the standard deviation obtained previously for the BaTiO3 NPs. Moreover, the SH spectra acquired before compression, after compression and with a π amplitude step function are in good agreement with the simulated results (Figure 4a).
Next, we studied the Au NPs. Au NPs are known to undergo both SH generation26,27 and two-photon absorption, giving rise to two-photon photoluminescence (TPPL).28 The onset of TPPL emission spectrally overlaps with the long wavelength part of the SH spectrum. TPPL from Au has been successfully used for mapping the plasmonic resonances of Au NPs.29 However, in the present case, the incoherent TPPL is rather a background contribution that might disturb precise measurements of the SH spectrum and therefore of the spectral phase. In qualitative agreement with a recent work by Deng et al.,30 we found that the ratio between the SH and TPPL is larger for Au dimers than for single 100-nm Au NPs. Moreover, the TPPL emission changes as a function of the spectral phase, as expected for a nonlinear process, being maximal for fully compressed laser pulses. As shown in Figure 4c for a single Au NP, no clear SH could be measured under the typical acquisition settings without compressing the laser pulse. To achieve a good signal-to-noise ratio for the SH signal, an integration time of at least 5 s was required. Nevertheless, even for this particle, regardless of the long integration time and the very high TPPL contribution partly overlapping with the SH signal, the measured spectral phase differed by no more than 0.2 rad from that obtained for the BaTiO3 NPs and the Au dimer (Figure 4e). Moreover, the SH spectrum could still be accurately controlled in the case of the Au dimer (Figure 4b) and, to a good extent, for the single NP as well (Figure 4c).
These observations show that the phase responses of all of the NPs investigated were the same: i.e., there is no additional phase information associated with the NPs. A priori, this result is expected for dielectric materials, such as BaTiO3 and Fe(IO3)3, that are transparent in our fundamental spectral range, whereas this is not necessarily the case for Au NPs. Indeed, it is well known that metallic NPs can show plasmonic resonances in the visible and near-infrared spectrum.31 Such resonances carry phase information that, in our experiment, might manifest as additional features in the phase measurement and would therefore affect the measurement of the laser spectral phase.32 Because our objective was to precisely measure the laser spectral phase, we chose Au NPs that were off-resonant with the excitation pulse, as confirmed by extinction measurements (data not shown), which showed that the plasmonic resonance was centered at ∼570 nm for the investigated NPs.
The results described above prove the independence of the phase measurement and SH control with respect to the NP size, shape and material. This conclusion directly impacts the ease of use of this method, as it shows that the NPs can reliably be used as local probes to measure minimal phase effects, without size and shape factors beyond fabrication control influencing the measurement. It is important to highlight that NPs can be adapted in size and material to best suit the sample under investigation. NPs of the size of a typical mode volume in a nanostructured sample, as well as biologically inert particles and particles with dielectric constants matched to the sample to minimize scattering, will reliably measure accurate nanoscopic nonlinear responses.
Conclusions
In this paper, we have shown that using the SH from individual NPs, in combination with a pulse shaper, allows one to obtain full phase and time control of a femtosecond laser pulse over a subdiffraction-limited area.
We have demonstrated this ability in two steps: first, using the SH from a single 150-nm NP, we were able to fully compress a femtosecond laser pulse in the time domain down to its Fourier limit using MIIPS; second, we showed that it is possible to deterministically control the SH emission from a single NP when applying specific spectral phases to the laser pulse. The universal character and robustness of our method are evident from the experiments reported for particles of different sizes, shapes and materials.
Taken together, these results demonstrate full femtosecond pulse control on a nanometer spatial scale and pave the way toward ultrafast and coherent control experiments on the nanoscale. In addition, the accuracy of phase control at the nanoscale achieved herein allows studies of the intrinsic phase response in nanostructures such as resonant plasmonic NPs. Finally, our SH control scheme allows SH NPs to be used as nanometer sources of tunable, coherent blue light.
References
Brinks D, Stefani FD, Kulzer F, Hildner R, Taminiau TH et al. Visualizing and controlling vibrational wave packets of single molecules. Nature 2010; 465: 905–908.
Hildner R, Brinks D, Nieder JB, Cogdell RJ, van Hulst NF . Quantum coherent energy transfer over varying pathways in single light-harvesting complexes. Science 2013; 340: 1448–1451.
Masia F, Accanto N, Langbein W, Borri P . Spin-flip limited exciton dephasing in CdSe/ZnS colloidal quantum dots. Phys Rev Lett 2012; 108: 087401.
Anderson A, Deryckx KS, Xu XG, Steinmeyer G, Raschke MB . Few-femtosecond plasmon dephasing of a single metallic nanostructure from optical response function reconstruction by interferometric frequency resolved optical gating. Nano Lett 2010; 10: 2519–2524.
Aeschlimann M, Brixner T, Fischer A, Kramer C, Melchior P et al. Coherent two-dimensional nanoscopy. Science 2011; 333: 1723–1726.
Biagioni P, Brida D, Huang JS, Kern J, Duò L et al. Dynamics of four-photon photoluminescence in gold nanoantennas. Nano Lett 2012; 12: 2941–2947.
Stockman MI . Ultrafast nanoplasmonics under coherent control. New J Phys 2008; 10: 025031.
Fuchs U, Zeitner U, Tünnermann A . Ultra-short pulse propagation in complex optical systems. Opt Express 2005; 13: 3852–3861.
Trebino R . Measuring the seemingly immeasurable. Nat Photon 2011; 5: 189–192.
Iaconis C, Walmsley IA . Spectral phase interferometry for direct electric-field reconstruction of ultrashort optical pulses. Opt Lett 1998; 23: 792–794.
Coello Y, Lozovoy VV, Gunaratne TC, Xu B, Borukhovich I et al. Interference without an interferometer: a different approach to measuring, compressing, and shaping ultrashort laser pulses. J Opt Soc Am B 2008; 25: A140–A150.
Xu B, Gunn JM, Cruz JM, Lozovoy VV, Dantus M . Quantitative investigation of the multiphoton intrapulse interference phase scan method for simultaneous phase measurement and compensation of femtosecond laser pulses. J Opt Soc Am B 2006; 23: 750–759.
Le Xuan L, Zhou C, Slablab A, Chauvat D, Tard C et al. Photostable second-harmonic generation from a single KTiOPO4 nanocrystal for nonlinear microscopy. Small 2008; 4: 1332–1336.
Extermann J, Bonacina L, Courvoisier F, Kiselev D, Mugnier Y et al. Nano-FROG: frequency resolved optical gating by a nanometric object. Opt Express 2008; 16: 10405–10411.
Li H, Zhang Z, Xu Q, Shi K, Jia Y et al. Characterizing ultrashort optical pulses using second-order nonlinear nanoprobes. Appl Phys Lett 2010; 97: 261108.
Wnuk P, Xuan LL, Slablab A, Tard C, Perruchas S et al. Coherent nonlinear emission from a single KTP nanoparticle with broadband femtosecond pulses. Opt Express 2009; 17: 4652–4658.
Aulbach J, Gjonaj B, Johnson P, Lagendijk A . Spatiotemporal focusing in opaque scattering media by wave front shaping with nonlinear feedback. Opt Express 2012; 20: 29237–29251.
Bäumner R, Bonacina L, Enderlein J, Extermann J, Fricke-Begemann T et al. Evanescent-field-induced second harmonic generation by noncentrosymmetric nanoparticles. Opt Express 2010; 18: 23218–23225.
Berweger S, Atkin JM, Xu XG, Olmon RL, Raschke MB . Femtosecond nanofocusing with full optical waveform control. Nano Lett 2011; 11: 4309–4313.
Neumann L, Pang Y, Houyou A, Juan ML, Gordon R et al. Extraordinary optical transmission brightens near-field fiber probe. Nano Lett 2011; 11: 355–360.
Staedler D, Magouroux T, Hadji R, Joulaud C, Extermann J et al. Harmonic nanocrystals for biolabeling: a survey of optical properties and biocompatibility. ACS Nano 2012; 6: 2542–2549.
Bidault S, Polman A . Water-based assembly and purification of plasmon-coupled gold nanoparticle dimers and trimers. Int J Opt 2012; 2012: 387274.
Brinks D, Hildner R, Stefani FD, van Hulst NF . Beating spatio-temporal coupling: implications for pulse shaping and coherent control experiments. Opt Express 2011; 19: 26486–26499.
McCabe DJ, Austin DR, Tajalli A, Weber S, Walmsley IA et al. Space–time coupling of shaped ultrafast ultraviolet pulses from an acousto-optic programmable dispersive filter. J Opt Soc Am B 2011; 28: 58–64.
Pestov D, Lozovoy VV, Dantus M . Multiple Independent Comb Shaping (MICS): phase-only generation of optical pulse sequences. Opt Express 2009; 17: 14351–14361.
Butet J, Duboisset J, Bachelier G, Russier-Antoine I, Benichou E et al. Optical second harmonic generation of single metallic nanoparticles embedded in a homogeneous medium. Nano Lett 2010; 10: 1717–1721.
Slablab A, Le Xuan L, Zielinski M, de Wilde Y, Jacques V et al. Second-harmonic generation from coupled plasmon modes in a single dimer of gold nanospheres. Opt Express 2012; 20: 220–227.
Castro-Lopez M, Brinks D, Sapienza R, van Hulst NF . Aluminum for nonlinear plasmonics: resonance-driven polarized luminescence of Al, Ag, and Au nanoantennas. Nano Lett 2011; 11: 4674–4678.
Ghenuche P, Cherukulappurath S, Taminiau TH, van Hulst NF, Quidant R . Spectroscopic mode mapping of resonant plasmon nanoantennas. Phys Rev Lett 2008; 101: 116805.
Deng HD, Li GC, Dai QF, Ouyang M, Lan S et al. Size dependent competition between second harmonic generation and two-photon luminescence observed in gold nanoparticles. Nanotechnology 2013; 24: 075201.
Novotny L, van Hulst N . Antennas for light. Nat Photon 2011; 5: 83–90.
Brinks D, Castro-Lopez M, Hildner R, van Hulst NF . Plasmonic antennas as design elements for coherent ultrafast nanophotonics. Proc Natl Acad Sci USA 2013; 110: 18386–18390. doi: 10.1073/pnas.1308652110.
Acknowledgements
This research was funded by the MICINN (programs Consolider Ingenio-2010: CSD2007-046-NanoLight.es, Plan Nacional FIS2009-0123: Optical NanoAntennas) and the European Union (ERC Advanced Grant 247330-NanoAntennas). LP acknowledges financial support from the Marie-Curie International Fellowship COFUND and ICFOnest program, and FP received support from the European Commission through the Erasmus Mundus Joint Doctorate Programme Europhotonics (Grant No. 159224-1-2009-1-FR-ERA MUNDUS-EMJD). DB acknowledges support from a Rubicon Grant of the Netherlands Organization for Scientific Research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Accanto, N., Nieder, J., Piatkowski, L. et al. Phase control of femtosecond pulses on the nanoscale using second harmonic nanoparticles. Light Sci Appl 3, e143 (2014). https://doi.org/10.1038/lsa.2014.24
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/lsa.2014.24
Keywords
This article is cited by
-
Nonlinear plasmonics: second-harmonic generation and multiphoton photoluminescence
PhotoniX (2023)
-
In situ electrical and thermal monitoring of printed electronics by two-photon mapping
Scientific Reports (2017)
