
Abstract
The World Health Organization (WHO) classification system has recently strengthened the diagnostic criteria for essential thrombocythemia (ET) by lowering the threshold platelet count, underscoring its morphological distinction from early/prefibrotic myelofibrosis (MF) and incorporating molecular markers of clonality. The International Working Group for Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) examined the clinical relevance of this process in 1104 cases of locally diagnosed ‘ET’ and showed worse overall, leukemia-free and fibrosis-free survival, and a higher risk of bleeding in early/prefibrotic MF (n=180) vs WHO-defined ET (n=891). The risk of thrombosis was similar between the two entities and, in WHO-defined ET, was predicted by thrombosis history, older age, cardiovascular risk factors and JAK2V617F. A prognostic model based on these risk factors identified patient groups in ET with residual risk of thrombosis, despite treatment with conventional therapy. The main objectives of the current perspective are to underscore the prognostic importance of morphological confirmation in the diagnosis of ET and provide management recommendations, in both WHO-defined ET and early/prefibrotic MF, based on observations from the aforementioned IWG-MRT and other studies. In so doing, we are fully cognizant and sympathetic of the fact that some of our recommendations need to be tested in prospective controlled studies.
Similar content being viewed by others


Introduction
Essential thrombocythemia (ET), polycythemia vera (PV) and primary myelofibrosis (PMF) are operationally classified as BCR-ABL1-negative myeloproliferative neoplasms (MPN).1 All three are believed to originate from a genetically transformed stem cell, which leads to clonal myeloproliferation. Neither the disease-initiating nor leukemia-promoting events in BCR-ABL1-negative MPN are known, although the majority of the patients harbor JAK2V617F or other secondary somatic mutations.2 In terms of diagnosis, the World Health Organization (WHO) classification system for hematopoietic tumors considers clonal erythrocytosis as being specific to PV, and uses bone marrow morphology to distinguish between ET and PMF.3
Distinguishing ET from early/prefibrotic PMF
Morphology
Diagnosis of WHO-defined ET requires absence of BCR-ABL1 (to exclude the possibility of ET-like chronic myeloid leukemia),4 absence of dyserythropoiesis (to exclude the possibility of myelodysplastic or overlap syndromes associated with thrombocytosis)5 and, most importantly, absence of bone marrow morphological changes that are more consistent with early/prefibrotic myelofibrosis (MF; megakaryocytes in ET are large, hyperlobulated and appear mature, and are usually not accompanied by erythroid or granulocyte proliferation, whereas those of early/prefibrotic MF display abnormal maturation with hyperchromatic, irregularly folded bulky nuclei and often accompanied by left-shifted granulocyte proliferation).6
In clinical practice, clues for a diagnosis of early/prefibrotic MF, in a patient suspected of having ET, include anemia, leukocytosis, increased serum lactate dehydrogenase level, leukoerythroblastosis and palpable splenomegaly.7 Some studies have also associated higher JAK2V617F allele burden with early/prefibrotic MF; in one study, no ET patient displayed an allele burden of >40%, whereas nearly 25% of patients with early/prefibrotic MF displayed >50% allele burden.8
Clinical relevance
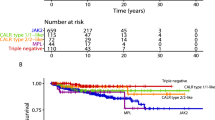
The International Working Group for MPN Research and Treatment (IWG-MRT) recently subjected 1104 cases of locally diagnosed ET to a central pathology review; 891 met the revised WHO criteria for ET and 180 for early/prefibrotic MF.9 The results revealed significantly worse overall survival (15-year survival 59% vs 80%), leukemic transformation rate (risk over 15 years 11.7% vs 2.1%) and fibrotic progression (risk over 15 years 16.9% vs 9.3%), in early/prefibrotic MF compared with WHO-defined ET.9 Patients with early/prefibrotic MF also suffered from a higher risk of bleeding (12% vs 6% incidence after 6–7 years follow-up), especially in the presence of aspirin therapy, leukocytosis or previous bleeding event.10 On the other hand, their thrombosis risk (10-year cumulative incidence of 17.9%)9 was similar to that of ET (10-year cumulative incidence of 16.2%),9 and risk factors for arterial thrombosis in early/prefibrotic MF included leukocytosis; overall thrombosis risk was lower in the presence of extreme thrombocytosis.11
Impact on clinical practice
On the basis of the above fact, we reiterate the prognostic importance of confirming the diagnosis of true ET in any patient with a reported diagnosis of ‘ET’. In this regard, one must carefully examine the complete blood count (anemia or leukocytosis suggest early/prefibrotic MF), peripheral blood smear (increased red cell distribution width, decreased mean corpuscular volume that is not associated with iron deficiency, or leukoerythroblastosis suggests early/prefibrotic MF), serum lactate dehydrogenase (increased levels are more consistent with early/prefibrotic MF) and JAK2V617F allele burden (diagnosis of ET must be questioned in the presence of >25% allele burden).
In terms of management, caution is required in the use of aspirin in early/prefibrotic MF because of the increased baseline risk of bleeding. In general, we recommend avoidance of prophylactic aspirin therapy in early/prefibrotic MF, especially in the presence of a bleeding history. This is different from our treatment approach in WHO-defined ET, in which we recommend universal use of aspirin therapy in the absence of clear contraindications, such as clinically relevant acquired von Willebrand syndrome. On the other hand, the indications to use cytoreductive therapy remain similar to those of ET, but might require deeper cytoreduction in the presence of leukocytosis.
WHO-defined ET
Survival and disease progression
In WHO-defined ET, life-expectancy is near-normal and the risk of leukemic or fibrotic progression, in the first 10 years of diagnosis, is <1%.9 This is particularly true in young patients without leukocytosis or history of thrombosis; the IWG-MRT has identified age ⩾60 years (2 points), leukocyte count ⩾11 × 109/l (1 point) and history of thrombosis (1 point) as independent predictors of poor survival in WHO-defined ET.12 The International Prognostic Score for ET (IPSET) uses these three risk factors and classifies patients into low (no adverse points), intermediate (1 or 2 adverse points) and high-(3 or more adverse points) risk categories, with corresponding median survivals of ‘not reached’, 25 years and 14 years.12 The IWG-MRT studies have also identified platelet count of >1000 × 109/l and thrombosis history as predictors of leukemic transformation, and absence of JAK2V617F, older age and anemia as predictors of progression into overt MF.9
Thrombosis and bleeding
The traditionally well-established risk factors for thrombosis in ET are advanced age and history of thrombosis. These two variables are conventionally used to delineate low (age <60 years and without thrombosis history) and high-(either age ⩾60 years or presence of thrombosis history) risk patients, in terms of thrombosis risk.13 Others have sometimes considered extreme thrombocytosis (platelet count >1000 or 1500 × 109/l) as a risk factor for thrombosis, but this was proven inaccurate by the IWG-MRT studies that have shown instead a reduced risk of arterial thrombosis in platelet millionaires with ET.14
Despite conventional risk-adapted therapy, there appears to be a residual risk of thrombosis in ET patients. In a series of publications, the IWG-MRT has identified age ⩾60 years, thrombosis history, cardiovascular risk factors, leukocyte count >11 × 109/l and presence of JAK2V617F as independent predictors of arterial thrombosis, and male gender as a predictor of venous thrombosis.14 The same set of variables, except leukocytosis, remained significant in the context of JAK2V617F-positive ET. In a subsequent analysis, the IWG-MRT developed a prognostic model for thrombosis in general (IPSET-thrombosis) by using four independent risk factors and hazard ratios from multivariable analysis as follows: age ⩾60 years (1 point), thrombosis history (2 points), cardiovascular risk factors (1 point), and JAK2V617F (2 points).15 Thrombosis risk was 1.03%/year in low (0 or 1 point), 2.35%/year in intermediate (2 points) and 3.56% in high-(3 or more points) risk patients.15
In yet another IWG-MRT study, bleeding complications in WHO-defined ET were more likely to occur in patients who are older, have experienced previous bleeding or are receiving aspirin therapy.10 Extreme thrombocytosis, in the absence of aspirin therapy, was not associated with excess bleeding.10
Impact on clinical practice
It is comforting for both patients and their physicians to recognize the outstanding prognosis associated with IPSET low or intermediate risk ET.12 Such patients are unlikely to benefit from participation in currently available clinical trials. The same can be said for high-risk patients with ET, unless the investigational drug under consideration harbors disease-modifying activity.
On the other hand, the findings from the IWG-MRT studies suggest a substantial amount of residual thrombosis risk in otherwise ‘appropriately’ treated patients with WHO-defined ET. This was most apparent in the presence of thrombosis history, in older patients with either cardiovascular risk factors or JAK2V617F, and in younger patients with both cardiovascular risk factors and JAK2V617F.15 By contrast, bleeding complications were less frequent but clearly exacerbated by aspirin therapy,10 whereas extreme thrombocytosis per se was not detrimental to either thrombosis or bleeding.10, 14
In view of the relatively high residual risk of thrombosis, in certain groups of patients with WHO-defined ET, it is reasonable to re-examine our current treatment approaches and consider some modifications. For example, further optimization of anti-platelet therapy is possible by changing the schedule of low-dose aspirin therapy from once-daily to twice-daily regimen,16 based on the observation that such modification overcomes biochemical resistance to aspirin.17 In other words, aspirin-induced inhibition of platelet function at 24 h might be incomplete in ET because of increased platelet turnover.18 This phenomenon is also seen in individuals with cardiovascular risk factors,19, 20 and has been associated with increased risk of arterial events.21 Regardless, the clinical relevance of reversing biochemical resistance to aspirin, both in the presence and absence of concomitant cytoreductive therapy, and the safety of twice-daily aspirin dosing must be evaluated in a controlled setting. Intensification of cytoreductive therapy constitutes another therapeutic option with the potential to further reduce the risk of recurrent thrombosis, and such an approach was recently shown to favorably affect thrombosis risk in PV.22
Treatment recommendations
Figure 1 summarizes our current treatment algorithm in WHO-defined ET, which takes into account the observations from the above-described IWG-MRT and other studies. In this regard, we find it practically useful to consider three layers of classification based on thrombosis history (no thrombosis vs arterial thrombosis vs venous thrombosis), age (<60 years vs older) and presence or absence of cardiovascular risk factors or JAK2V617F.

Contemporary treatment algorithm in essential thrombocythemia. CVR, cardiovascular risk factors.
Observation alone is acceptable in asymptomatic young patients without thrombosis history, cardiovascular risk factors or JAK2V617F.23 Such patients should avoid aspirin use in the presence of platelet count >1000 × 109/l (aspirin therapy in such cases might increase bleeding risk).23 The presence of microvascular symptoms, cardiovascular risk factors or JAK2V617F in young, otherwise low-risk patients with ET justifies the use of once-daily aspirin therapy;24 in at least one study, the presence of cardiovascular symptoms or JAK2V617F was associated with increased risk of venous and arterial thrombosis, respectively.23 Persistence of microvascular symptoms despite once-daily aspirin dosing or presence of both cardiovascular risk factors and JAK2V617F is a reasonable rationale to consider using twice-daily aspirin dosing, provided platelet count is <1000 × 109/l.
Thrombosis history in young patients with ET mandates cytoreductive therapy. Our first-line drug of choice, in this regard, is hydroxyurea and second-line drug of choice pegylated interferon. The goal would be to adequately control myeloproliferation and keep leukocyte and platelet counts in the normal range. In addition, patients with venous thrombosis might require systemic anticoagulation, whereas those with arterial events are treated with once-daily aspirin. The duration of systemic anticoagulation depends on whether or not the venous event was provoked (6 months) or unprovoked (indefinite). ‘Provoked’ in this instance includes inadequately treated disease. Because cytoreductive therapy ameliorates the increased platelet turnover associated with ET, twice-daily aspirin therapy might not be necessary in this instance, even in the presence of cardiovascular risk factors or JAK2V617F.
Older patients (age ⩾60 years) with or without thrombosis history are usually offered treatment with both hydroxyurea and at least once-daily aspirin. However, the evidence for the value of cytoreductive therapy, in the absence of all risk factors (thrombosis history, cardiovascular risk factors and JAK2V617F) other than age, is not strong enough to discourage the use of aspirin only in certain circumstances. For example, an otherwise asymptomatic patient with ET who has been treated with aspirin only for many years may not require institution of cytoreductive therapy just because he or she turned 60.
Older patients with venous thrombosis history require systemic anticoagulation, in addition to cytoreductive therapy. As stated above for younger patients, the duration of systemic anticoagulation depends on whether or not the venous event was provoked (6 months) or unprovoked (indefinite). Such patients should also receive once-daily aspirin therapy in the presence of cardiovascular risk factors or JAK2V617F, and in the absence contraindications to aspirin use. Older patients with arterial thrombosis do not require systemic anticoagulation. However, such patients might benefit from twice-daily aspirin dosing, in addition to cytoreductive therapy, in the presence of cardiovascular risk factors or JAK2V617F. In older patients with ET, busulfan or pegylated interferon is a reasonable alternative in case of hydroxyurea intolerance or resistance.
Concluding remarks
The main objectives of the current perspective are to underscore the prognostic relevance of distinguishing WHO-defined ET from early/prefibrotic MF and to increase awareness regarding the relatively high risk of residual thrombosis in certain group of patients with WHO-defined ET. We believe that these issues constitute actionable challenges and need to be ultimately evaluated in prospective controlled studies. Our recommendations outlined in Figure 1 are neither binding nor absolute. They are simply there to assist those who seek our opinion.
References
Tefferi A, Vardiman JW . Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia 2008; 22: 14–22.
Tefferi A . Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia 2010; 24: 1128–1138.
Barbui T, Thiele J, Vannucchi AM, Tefferi A . Problems and pitfalls regarding WHO-defined diagnosis of early/prefibrotic primary myelofibrosis versus essential thrombocythemia. Leukemia 2013; e-pub ahead of print 7 March 2013; doi: 10.1038/leu.2013.74.
Michiels JJ, Berneman Z, Schroyens W, Kutti J, Swolin B, Ridell B et al. Philadelphia (Ph) chromosome-positive thrombocythemia without features of chronic myeloid leukemia in peripheral blood: natural history and diagnostic differentiation from Ph-negative essential thrombocythemia. Ann Hematol 2004; 83: 504–512.
Boissinot M, Garand R, Hamidou M, Hermouet S . The JAK2-V617F mutation and essential thrombocythemia features in a subset of patients with refractory anemia with ring sideroblasts (RARS). Blood 2006; 108: 1781–1782.
Kvasnicka HM, Thiele J . Prodromal myeloproliferative neoplasms: the 2008 WHO classification. Am J Hematol 2010; 85: 62–69.
Carobbio A, Finazzi G, Thiele J, Kvasnicka HM, Passamonti F, Rumi E et al. Blood tests may predict early primary myelofibrosis in patients presenting with essential thrombocythemia. Am J Hematol 2012; 87: 203–204.
Hussein K, Bock O, Theophile K, von Neuhoff N, Buhr T, Schlue J et al. JAK2(V617F) allele burden discriminates essential thrombocythemia from a subset of prefibrotic-stage primary myelofibrosis. Exp Hematol 2009; 37: 1186–1193 e1187.
Barbui T, Thiele J, Passamonti F, Rumi E, Boveri E, Ruggeri M et al. Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: an international study. J Clin Oncol 2011; 29: 3179–3184.
Finazzi G, Carobbio A, Thiele J, Passamonti F, Rumi E, Ruggeri M et al. Incidence and risk factors for bleeding in 1104 patients with essential thrombocythemia or prefibrotic myelofibrosis diagnosed according to the 2008 WHO criteria. Leukemia 2012; 26: 716–719.
Buxhofer-Ausch V, Gisslinger H, Thiele J, Gisslinger B, Kvasnicka HM, Mullauer L et al. Leukocytosis as an important risk factor for arterial thrombosis in WHO-defined early/prefibrotic myelofibrosis: an international study of 264 patients. Am J Hematol 2012; 87: 669–672.
Passamonti F, Thiele J, Girodon F, Rumi E, Carobbio A, Gisslinger H et al. A prognostic model to predict survival in 867 World Health Organization-defined essential thrombocythemia at diagnosis: a study by the International Working Group on Myelofibrosis Research and Treatment. Blood 2012; 120: 1197–1201.
Tefferi A . Polycythemia vera and essential thrombocythemia: 2012 update on diagnosis, risk stratification, and management. Am J Hematol 2012; 87: 285–293.
Carobbio A, Thiele J, Passamonti F, Rumi E, Ruggeri M, Rodeghiero F et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood 2011; 117: 5857–5859.
Barbui T, Finazzi G, Carobbio A, Thiele J, Passamonti F, Rumi E et al. Development and validation of an International Prognostic Score of thrombosis in World Health Organization-essential thrombocythemia (IPSET-thrombosis). Blood 2012; 120: 5128–5133, ; quiz 5252.
Tefferi A . Overcoming "aspirin resistance" in MPN. Blood 2012; 119: 3377–3378.
Pascale S, Petrucci G, Dragani A, Habib A, Zaccardi F, Pagliaccia F et al. Aspirin-insensitive thromboxane biosynthesis in essential thrombocythemia is explained by accelerated renewal of the drug target. Blood 2012; 119: 3595–3603.
Di Minno MN, Lupoli R, Palmieri NM, Russolillo A, Buonauro A, Di Minno G . Aspirin resistance, platelet turnover, and diabetic angiopathy: a 2011 update. Thromb Res 2011; 129: 341–344.
Dragani A, Pascale S, Recchiuti A, Mattoscio D, Lattanzio S, Petrucci G et al. The contribution of cyclooxygenase-1 and -2 to persistent thromboxane biosynthesis in aspirin-treated essential thrombocythemia: implications for antiplatelet therapy. Blood 2010; 115: 1054–1061.
Grove EL, Hvas AM, Mortensen SB, Larsen SB, Kristensen SD . Effect of platelet turnover on whole blood platelet aggregation in patients with coronary artery disease. J Thromb Haemost 2011; 9: 185–191.
Snoep JD, Hovens MM, Eikenboom JC, van der Bom JG, Huisman MV . Association of laboratory-defined aspirin resistance with a higher risk of recurrent cardiovascular events: a systematic review and meta-analysis. Arch Intern Med 2007; 167: 1593–1599.
Marchioli R, Finazzi G, Specchia G, Cacciola R, Cavazzina R, Cilloni D et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med 2013; 368: 22–33.
Alvarez-Larran A, Cervantes F, Pereira A, Arellano-Rodrigo E, Perez-Andreu V, Hernandez-Boluda JC et al. Observation versus antiplatelet therapy as primary prophylaxis for thrombosis in low-risk essential thrombocythemia. Blood 2010; 116: 1205–1210, quiz 1387.
Barbui T . How to manage children and young adults with myeloproliferative neoplasms. Leukemia 2012; 26: 1452–1457.
Acknowledgements
We would like to formally acknowledge the superior contributions from the many investigators who participated in the International Working Group for Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) projects. Tiziano Barbui is supported by a AIRC 5 × 1000 grant of the AGIMM group.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Tefferi, A., Barbui, T. Personalized management of essential thrombocythemia—application of recent evidence to clinical practice. Leukemia 27, 1617–1620 (2013). https://doi.org/10.1038/leu.2013.99
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2013.99
Keywords
This article is cited by
-
Cardiovascular Safety of Anagrelide Hydrochloride versus Hydroxyurea in Essential Thrombocythaemia
Cardiovascular Toxicology (2021)
-
Hematological Malignancies and Arterial Thromboembolism
Indian Journal of Hematology and Blood Transfusion (2019)
-
Risk Factors for and Management of MPN-Associated Bleeding and Thrombosis
Current Hematologic Malignancy Reports (2017)
-
Oral anticoagulation to prevent thrombosis recurrence in polycythemia vera and essential thrombocythemia
Annals of Hematology (2015)
-
An overview on CALR and CSF3R mutations and a proposal for revision of WHO diagnostic criteria for myeloproliferative neoplasms
Leukemia (2014)
