
Abstract
In 1951, William Dameshek described the concept of ‘myeloproliferative disorders (MPDs)’ by grouping together chronic myelogenous leukemia (CML), polycythemia vera (PV), essential thrombocythemia (ET), primary myelofibrosis (PMF) and erythroleukemia; he reasoned that a self-perpetuating trilineage myeloproliferation underlined their pathogenesis. Pre-Dameshek luminaries who laid the foundation for this unifying concept include Bennett, Virchow, Heuck, Vaquez, Osler, Di Guglielmo and Epstein. In 1960, Nowell and Hungerford discovered the Philadelphia (Ph) chromosome in CML. In 1967, Fialkow and colleagues used X-linked polymorphisms to establish CML as a clonal stem cell disease. Also in 1967, the PV Study Group was summoned by Louis Wasserman to study the natural history of PV and conduct large-scale clinical trials. In 1972, Janet Rowley deciphered the Ph chromosome as a reciprocal translocation between chromosomes 9 and 22, thus paving the way for its subsequent characterization as an oncogenic BCR–ABL mutation. In 1996, Brian Druker discovered imatinib—a small molecule ABL inhibitor with exceptional therapeutic activity in CML. In 2005, a gain-of-function JAK2 mutation (JAK2V617F) was described in BCR–ABL-negative MPDs, raising the prospect of a CML-like treatment strategy in PV, ET and PMF. The current review considers these and other landmark events in the history of MPDs.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others
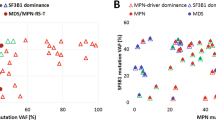
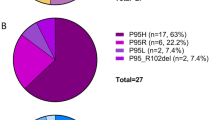
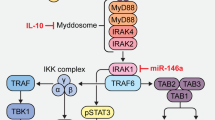
References
Bennett JH . Case of hypertrophy of the spleen and liver in which death took place from suppuration of the blood. Edinb Med Surg J 1845; 64: 413–423.
Virchow R . Weisses Blut. Froriep's Notzien 1845; 36: 151–156.
Craigie D . Case of disease and enlargement of the spleen in which death took place from the presence of purulent matter in the blood. Edinb Med Surg J 1845; 64: 400–413.
Vaquez H . Sur une forme speciale de cyanose s'accompanant d'hyperglobulie excessive et peristente (On a special form of cyanosis accompanied by excessive and persistent erythrocytosis). Compt rend Soc de biol and suppl note, Bull et mem Soc med d'hop de Paris, 3 ser 1895; 12: 60 1892; 4: 384–388.
Osler W . Chronic cyanosis, with polycythemia and enlarged spleen: a new clinical entity. Am J Med Sci 1903; 126: 187–201.
Heuck G . Zwei Falle von Leukamie mit eigenthumlichem Blut- resp. Knochenmarksbefund (Two cases of leukemia with peculiar blood and bone marrow findings, respectively). Arch Pathol Anat Physiol Virchows 1879; 78: 475–496.
Epstein E, Goedel A . Hamorrhagische thrombozythamie bei vascularer schrumpfmilz (Hemorrhagic thrombocythemia with a vascular, sclerotic spleen). Virchows Archiv A Pathol Anat Histopathol 1934; 293: 233–248.
Dameshek W . Some speculations on the myeloproliferative syndromes. Blood 1951; 6: 372–375.
Groffen J, Stephenson JR, Heisterkamp N, de Klein A, Bartram CR, Grosveld G . Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell 1984; 36: 93–99.
Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005; 7: 387–397.
James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005; 434: 1144–1148.
Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005; 352: 1779–1790.
Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005; 365: 1054–1061.
Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med 2007; 356: 459–468.
Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 2006; 3: e270.
Nowell PC, Hungerford DA . Chromosome studies on normal and leukemic human leukocytes. J Natl Cancer Inst 1960; 25: 85–109.
Rowley JD . Letter: a new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973; 243: 290–293.
de Klein A, van Kessel AG, Grosveld G, Bartram CR, Hagemeijer A, Bootsma D et al. A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukaemia. Nature 1982; 300: 765–767.
Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr–Abl positive cells. Nat Med 1996; 2: 561–566.
Cruse JM . History of medicine: the metamorphosis of scientific medicine in the ever-present past. Am J Med Sci 1999; 318: 171–180.
Falagas ME, Zarkadoulia EA, Bliziotis IA, Samonis G . Science in Greece: from the age of Hippocrates to the age of the genome. FASEB J 2006; 20: 1946–1950.
Christie RV . Galen on Erasistratus. Perspect Biol Med 1987; 30: 440–449.
Sigerist HE . A History of Medicine. Oxford University press: New York, 1961; 1: 564.
Fahraeus R . The suspension stability of blood. Acta Med Scand 1921; 55: 1–228.
Avicenna . A treatise on the'Canon of Medicine' of Avicenna (980–1037). Luzac & Co: London, 1930, 276.
Simon SR . Moses Maimonides: medieval physician and scholar. Arch Intern Med 1999; 159: 1841–1845.
Lieber E . Galen: physician as philosopher; Maimonides: philosopher as physician. Bull Hist Med 1979; 53: 268–285.
Schmidt PJ . Transfuse George Washington!. Transfusion 2002; 42: 275–277.
Harvey W . Exercitatio Anatomica de Motu Cordis et Sanguinis in Animalibus. 1628.
Fleming D . Galen on the motions of the blood in the heart and lungs. Isis 1955; 46: 14–21.
Hooke R . Micrographia: Or, Some Physiological Descriptions of Minute Bodies Made by Magnifying Glasses'. London, J Martyn and J Allestry: London, (1st edn), 1665.
van Leeuwenhoek A . Philosophical transactions of the Royal Society. London 1674; 9: 121–128.
Cobb M . Reading and Writing The Book of Nature: Jan Swammerdam (1637–1680). Endeavour 2000; 24: 122–128.
Lieutaud J . Elementa Physiologiae. Amsterdam (translated and quoted in Dryefus, C 1957, Milestones in the History of Haematology, pp 11–12, New York) 1749, 82–84.
Gulliver G . The works of William Hewson. FRS London, The Sydenham Society 1846; part III, 1vi: 360pp, p 214.
Doyle D . William Hewson (1739–74): the father of haematology. Br J Haematol 2006; 133: 375–381.
Addison W . Colorless globules in the buffy coat of the blood. Lond Med Gaz NS 1840, 1841 27: 477–479.
Donne A . De l'origine des globules du sang, de leur mode de formation et de leur fin. Comptes rendus de l'Académie des Sciences, Paris 1842; 14: 168–366.
Brewer DB . Max Schultze (1865), G. Bizzozero (1882) and the discovery of the platelet. Br J Haematol 2006; 133: 251–258.
Ehrlich P . Beitrag zur Kenntnis der Anilinfarbungen under ihrer Verwendung in der Microscopischen Technik. Archives Mikrochirurgie Anatomischer 1877; 13: 263–277.
Drews J . Paul Ehrlich: magister mundi. Nat Rev Drug Discov 2004; 3: 797–801.
Piller G . Leukaemia—a brief historical review from ancient times to 1950. Br J Haematol 2001; 112: 282–292.
Neumann E . Ueber die Bedeutung des Knochenmarkes fur die Blutbildung. Ein Beitrag zur Entwicklungsgeschichte der Blutkorperchen. Archives Heilkunde 1869; 10: 68–102.
Ehrlich P . Uber die Bedeutung der neutrophilen Kornung. Charite Annales 1887; 12: 288–295.
Askanazy M . Ueber extrauterine Bildung von Blutzellen in der Leber. Verh Dtsch Pathol Ges 1904; 7: 58–65.
Assmann H . Beitrage zur osteosklerotischen anamie. Beitr Pathol Anat Allgemeinen Pathologie (Jena) 1907; 41: 565–595.
Meyer E, Heinke A . Uber Blutbildung bei schweren Anamien und Leukamien. Dtsch Arch Klin Med 1907; 88: 435–492.
Donhauser J . The human spleen as an haematoplastic organ, as exemplified in a case of splenomegaly with sclerosis of the bone-marrow. J Exp Med 1908; 10: 559–574.
Hirschfeld H . Die generalisierte aleukamische Myelose und ihre Stellung im System der leukamischen Erkrankungen. Z Klin Med 1914; 80: 126–173.
Jackson H Jr, Parker F Jr, Lemon HM . Agnogenic myeloid metaplasia of the spleen: a syndrome simulating other more definite hematological disorders. N Engl J Med 1940; 222: 985–994.
Jaffe ES, Harris NL, Stein H, Vardiman JW . World Health Organization Classification of Tumours of Hematopoietic and Lymphoid Tissues. IARC Press: Lyon, France, 2001, 1–351.
Mesa RA, Verstovsek S, Cervantes F, Barosi G, Reilly JT, Dupriez B et al. Primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (post-PV MF), post essential thrombocythemia myelofibrosis (post-ET MF), blast phase PMF (PMF-BP): Consensus on terminology by the international working group for myelofibrosis research and treatment (IWG-MRT). Leuk Res 2007; 31: 737–740.
Hirsch R . Generalized osteosclerosis with chronic polycythemia vera. Arch Pathol 1935; 19: 91–97.
Vaughan JM, Harrison CV . Leuco-erythrobalstic anaemia and myelosclerosis. J Pathol Bacteriol 1939; 48: 339–352.
Rosenthal N, Erf LA . Clinical observations on osteopetrosis and myelofibrosis. Arch Intern Med 1943; 71: 793–813.
Heller EL, Lewisohn MG, Palin WE . Aleukemic myelosis: chronic nonleukemic myelosis, agnogenic myeloid metaplasia, osteosclerosis, leuko-erythroblastic anemia, and synonymous designations. Am J Pathol 1947; 23: 327–365.
Silverstein MN . Agnogenic myeloid metaplasia. 1975, 126.
Cabot RC . A case of chronic cyanosis without discernible cause, ending in cerebral hemorrhage. Boston Med Surg J 1899; 141: 574–575.
Cabot RC . A second case of chronic cyanosis without assignable cause. Boston Med Sug J 1900; 142: 275.
McKeen SF . A case of marked cyanosis, difficult to explain. Boston Med Surg J 1901; 144: 610.
Saundby R, Russell AE . An unexplained condition of chronic cyanosis, with a report of a case. Lancet 1902; 1: 515.
Robb-Smith AHT . Osler's influence on haematology. Blood Cells 1981; 7: 513–533.
Osler W . A clinical lecture on erythraemia (polycythaemia with cyanosis, maladie de Vaquez). Lancet 1908; 1: 143–146.
Turk W . Beitrage zur kenntnis des symptomenbildes polyzythamie mit milztumor und zyanose. Wiener medizinische Wochenschrift 1904; 17: 153–160, 189–193.
Weber FP, Watson JH . Chronic polycythemia with enlarged spleen, probably a disease of the bone marrow. Trans Clin Soc 1904; 37: 115.
Parkes-Weber F . Polycythemia, erythrocytosis and erythraemia. QJM 1908; 2: 85–134.
Di Guglielmo G . Richerche di ematologia. I. Un caso di eritroleucemia. Megacariociti in circolo e loro funzione piastrinopoietico. Folia Med (Pavia) 1917; 13: 386.
Di Guglielmo G . Eritremie acute. Bolletino della Societa di Medicina e Chirugia di pavia 1923; 40: 665.
Di Guglielmo G . Myelosi eritremica cronica. Haematologica 1942; 24: 1–58.
Martin WJ, Bayrd ED . Erythroleukemia, with special emphasis on the acute or incomplete variety; report of five cases. Blood 1954; 9: 321–339.
Vardiman JW, Harris NL, Brunning RD . The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 2002; 100: 2292–2302.
Di Guglielmo G . Eritroleucemia e pistrinemia. Folia Med 1920; 1: 36.
Fanger H, Cella Jr LJ, Litchman H . Thrombocythemia; report of three cases and review of literature. N Engl J Med 1954; 250: 456–461.
Gunz FW . Hemorrhagic thrombocythemia: a critical review. Blood 1960; 15: 706–723.
Ozer FL, Traux WE, Miesch DC, Levin WC . Primary hemorrhagic thrombocythemia. Am J Med 1960; 28: 807–823.
Gunz FW . William Dameshek, 1900–1969. Blood 1970; 35: 577–582.
Stratton HM . William Dameshek, a personal remembrance. Blood 1970; 35: 4–5.
Jaffe ER . Commentary: the evolution of the American and International Societies of Hematology: a brief history. Am J Hematol 1992; 39: 67–69.
Tullis JL . William Dameshek, 1900–1969. Blood 1970; 35: 1–3.
Parkes-Weber F, Watson JH . Chronic polycythemia with enlarged spleen, probably a disease of the bone marrow. Br J Med 1904; 1: 729.
Pianese G . Per una miglior conoscenza dei megacariociti. Haematologica 1920; 1: 61–110.
Minot GR, Buckman TE . Erythremia (polycythemia rubra vera). The development of anemia; the relation to leukemia; consideration of the basal metabolism, blood formation and destruction and fragility of the red cells. Am J Med Sci 1923; 166: 469–489.
Nowell PC . Progress with chronic myelogenous leukemia: a personal perspective over four decades. Annu Rev Med 2002; 53: 1–13.
Hsu TC, Pomerat CM . Mammalian chromosomes in vitro II. A method for spreading the chromosomes of cells in tissue culture. J Hered 1953; 44: 23–29.
Nowell PC, Hungerford DA . A minute chromosome in human granulocytic leukemia. Science 1960; 132: 1497.
Tjio J-H, Levan A . The chromosome number of man. Hereditas 1956; 42: 1–6.
Lejeune J, Gautier M, Turpin R . Etude des chromosomes somatiques de neuf enfants mongoliens. CR Seances Acad Sci 1959; 248: 1721–1722.
Boveri T . Zur Frage der Entstehung maligner Tumoren. Gustav Fischer, Jena 1914.
Beutler E, Yeh M, Fairbanks VF . The normal human female as a mosaic of X-chromosome activity: studies using the gene for C-6-PD-deficiency as a marker. Proc Natl Acad Sci USA 1962; 48: 9–16.
Lyon MF . Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 1961; 190: 372–373.
Ohno S, Makino S . The single-X nature of sex chromatin in man. Lancet 1961; 1: 78–79.
Ohno S, Hauschka TS . Allocycly of the X-chromosome in tumors and normal tissues. Cancer Res 1960; 20: 541–545.
Linder D, Gartler SM . Glucose-6-phosphate dehydrogenase mosaicism: utilization as a cell marker in the study of leiomyomas. Science 1965; 150: 67–69.
Fialkow PJ, Gartler SM, Yoshida A . Clonal origin of chronic myelocytic leukemia in man. Proc Natl Acad Sci USA 1967; 58: 1468–1471.
Adamson JW, Fialkow PJ, Murphy S, Prchal JF, Steinmann L . Polycythemia vera: stem-cell and probable clonal origin of the disease. N Engl J Med 1976; 295: 913–916.
Jacobson RJ, Salo A, Fialkow PJ . Agnogenic myeloid metaplasia: a clonal proliferation of hematopoietic stem cells with secondary myelofibrosis. Blood 1978; 51: 189–194.
Fialkow PJ, Faguet GB, Jacobson RJ, Vaidya K, Murphy S . Evidence that essential thrombocythemia is a clonal disorder with origin in a multipotent stem cell. Blood 1981; 58: 916–919.
Martin PJ, Najfeld V, Hansen JA, Penfold GK, Jacobson RJ, Fialkow PJ . Involvement of the B-lymphoid system in chronic myelogenous leukaemia. Nature 1980; 287: 49–50.
Raskind WH, Jacobson R, Murphy S, Adamson JW, Fialkow PJ . Evidence for the involvement of B lymphoid cells in polycythemia vera and essential thrombocythemia. J Clin Invest 1985; 75: 1388–1390.
Reeder TL, Bailey RJ, Dewald GW, Tefferi A . Both B and T lymphocytes may be clonally involved in myelofibrosis with myeloid metaplasia. Blood 2003; 101: 1981–1983.
Tefferi A, Schad CR, Pruthi RK, Ahmann GJ, Spurbeck JL, Dewald GW . Fluorescent in situ hybridization studies of lymphocytes and neutrophils in chronic granulocytic leukemia. Cancer Genet Cytogenet 1995; 83: 61–64.
Buschle M, Janssen JW, Drexler H, Lyons J, Anger B, Bartram CR . Evidence for pluripotent stem cell origin of idiopathic myelofibrosis: clonal analysis of a case characterized by a N-ras gene mutation. Leukemia 1988; 2: 658–660.
Pardanani A, Lasho TL, Finke C, Markovic SN, Tefferi A . Demonstration of MPLW515K, but not JAK2V617F, in in vitro expanded CD4+ T lymphocytes. Leukemia advance online publication 17 May 2007; doi: 101038/sjleu2404749 2007.
Bellanne-Chantelot C, Chaumarel I, Labopin M, Bellanger F, Barbu V, De Toma C et al. Genetic and clinical implications of the Val617Phe JAK2 mutation in 72 families with myeloproliferative disorders. Blood 2006; 108: 346–352.
Raskind WH, Ferraris AM, Najfeld V, Jacobson RJ, Moohr JW, Fialkow PJ . Further evidence for the existence of a clonal Ph-negative stage in some cases of Ph-positive chronic myelocytic leukemia. Leukemia 1993; 7: 1163–1167.
Kovitz C, Kantarjian H, Garcia-Manero G, Abruzzo LV, Cortes J . Myelodysplastic syndromes and acute leukemia developing after imatinib mesylate therapy for chronic myeloid leukemia. Blood 2006; 108: 2811–2813.
Kralovics R, Teo SS, Li S, Theocharides A, Buser AS, Tichelli A et al. Acquisition of the V617F mutation of JAK2 is a late genetic event in a subset of patients with myeloproliferative disorders. Blood 2006; 108: 1377–1380.
Pardanani A, Lasho TL, Finke C, Mesa RA, Hogan WJ, Ketterling RP et al. Extending JAK2V617F and MPLW515 mutation analysis to single hematopoietic colonies and B- and T-lymphocytes. Stem Cells 2007; Published online June 1, 2007, DOI:10.1634/stemcells.2007-0175.
Harrison CN, Gale RE, Machin SJ, Linch DC . A large proportion of patients with a diagnosis of essential thrombocythemia do not have a clonal disorder and may be at lower risk of thrombotic complications. Blood 1999; 93: 417–424.
Antonioli E, Guglielmelli P, Pancrazzi A, Bogani C, Verrucci M, Ponziani V et al. Clinical implications of the JAK2 V617F mutation in essential thrombocythemia. Leukemia 2005; 19: 1847–1849.
Wasserman LR . Polycythemia Vera Study Group: a historical perspective. Semin Hematol 1986; 23: 183–187.
Tinney WS, Hall BE, Giffen HZ . Hematologic complications of P. Vera. Proc Staff Meet Mayo Clin 1943; 18: 227–230.
Hall BE . Therapeutic use of radiophosphorus in polycythemia vera, leukemia and allied diseases. In: The Use of Isotopes in Biology and Medicine. University of Wisconsin Press: WI, 1948, 353–376.
Ludin M . Ein betrag zur kentniss der symptmologie und therapie der primaren polycythemie. Z Klin Med 1917; 84: 460–476.
Eppinger H, Kloss K . Zur Therapie der Polyzythaemie. Therapeutis du Monatshefte 1918; 32: 322–326.
Forkner CE, Scott TFM, Wu SC . Treatment of polycythemia vera (erythremia) with a solution of potassium arsenite. Arch Intern Med 1933; 51: 616–629.
Lawrence JH . Nuclear physics and therapy: preliminary report on a new method for the treatment of leukemia and polycythemia vera. Radiology 1940; 35: 51–60.
Falconer EH . The treatment of polycythemia vera with lead compounds. Am J Med 1942; 203: 856–857.
Shullenberger CC, Watkins CH . Effects of nitrogen mustard on the bone marrow in polycythemia vera. Ann Intern Med 1950; 33: 841–853.
Rosenthal N, Rosenthal RL . Treatment of polycythemia vera with triethylene melamine: summary of thirty cases. Arch Intern Med 1952; 90: 379.
Isaacs R . Treatment of polycythemia vera with daraprim. J Am Med Assoc 1954; 156: 1491–1493.
Wald N, Hoshino T, Sears ME . Therapy of polycythemia vera with myleran. Blood 1958; 13: 757–762.
Shullenberger CC . Long-range treatment of polycythemia vera with 6-mercaptopurine. Cancer Chemother Rep 1962; 16: 251–252.
Bond WH . Evaluation of compound 8103 Abbott (N, N′ bis (bromopropionyl) piperazine) in the treatment of polycythemia rubra vera. Proc Am Assoc Cancer Res 1962; 3: 306.
Perkins J, Israuels MC, Wilkinson JF . Polycythaemia vera: clinical studies on a series of 127 patients managed without radiation therapy. QJM 1964; 33: 499–518.
Varela JE, Kremenchuzky S, Fraga A, Etcheverry MA, Figueiras H . Evaluation of haemopoiesis and comparative study of 32P and chlorambucil therapy by means of radiotracers in polycythemia vera. Minerva Nucl 1965; 9: 275–280.
Pengelly CD . Reduction of haematocrit and red-blood-cell volume in patients with polycythaemia secondary to hypoxic lung disease by dapsone and pyrimethamine. Lancet 1966; 2: 1381–1386.
West WO, Ruff JD, Yarboro JW . Response of polycythemia to treatment with a new agent: hydroxyurea (abstract). Ann Intern Med 1970; 72: 795.
Logue GL, Gutterman JU, McGinn TG, Laszlo J, Rundles RW . Melphalan therapy of polycythemia vera. Blood 1970; 36: 70–86.
Osler W . Chronic cyanosis with polycythemia and enlarged spleen: a new clinical entity. Am J Sci 1903; 126: 187–201.
Chievitz E, Thiede T . Complications and causes of death in polycythemia vera. Acta Med Scand 1962; 172: 513–523.
Berk PD, Wasserman LR, Fruchtman SM, Goldberg JD . Treatment of polycythemia vera: a summary of clinical trials conducted by the Polycythemia Vera Study Group. In: Wasserman LR, Berk PD, Berlin NI (eds). Polycythemia Vera and the Myeloproliferative Disorders. W.B. Saunders: Philadelphia, 1995, 166–194.
Fruchtman SM, Mack K, Kaplan ME, Peterson P, Berk PD, Wasserman LR . From efficacy to safety—a Polycythemia Vera Study Group report on hydroxyurea in patients with polycythemia vera. Semin Hematol 1997; 34: 17–23.
Tartaglia AP, Goldberg JD, Berk PD, Wasserman LR . Adverse effects of antiaggregating platelet therapy in the treatment of polycythemia vera. Semin Hematol 1986; 23: 172–176.
Landolfi R, Marchioli R, Kutti J, Gisslinger H, Tognoni G, Patrono C et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med 2004; 350: 114–124.
Tefferi A, Thiele J, Orazi A, Kvasnicka HM, Barbui T, Hanson CA et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. 10.1182/blood-2007-04-083501. Blood 2007: 110: 1092–1097.
Cortelazzo S, Finazzi G, Ruggeri M, Vestri O, Galli M, Rodeghiero F et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Eng J Med 1995; 332: 1132–1136.
Harrison CN, Campbell PJ, Buck G, Wheatley K, East CL, Bareford D et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med 2005; 353: 33–45.
Gangat N, Wolanskyj AP, McClure RF, Li CY, Schwager S, Wu W et al. Risk stratification for survival and leukemic transformation in essential thrombocythemia: a single institutional study of 605 patients. Leukemia 2007; 21: 270–276.
Finazzi G, Caruso V, Marchioli R, Capnist G, Chisesi T, Finelli C et al. Acute leukemia in polycythemia vera. An analysis of 1,638 patients enrolled in a prospective observational study. Blood 2005; 105: 2664–2670.
Berlin NI . Prologue—polycythemia vera—the closing of the Wasserman–Polycythemia Vera Study Group era. Semin Hematol 1997; 34: 1–5.
Seabright M . A rapid banding technique for human chromosomes. Lancet 1971; 2: 971–972.
Caspersson T, Zech L, Johansson C . Differential binding of alkylating fluorochromes in human chromosomes. Exp Cell Res 1970; 60: 315–319.
Caspersson T, Gahrton G, Lindsten J, Zech L . Identification of the Philadelphia chromosome as a number 22 by quinacrine mustard fluorescence analysis. Exp Cell Res 1970; 63: 238–240.
Rowley JD, Golomb HM, Dougherty C . 15/17 translocation, a consistent chromosomal change in acute promyelocytic leukaemia. Lancet 1977; 1: 549–550.
Rowley JD . Identificaton of a translocation with quinacrine fluorescence in a patient with acute leukemia. Ann Genet 1973; 16: 109–112.
Abelson HT, Rabstein LS . Lymphosarcoma: virus-induced thymic-independent disease in mice. Cancer Res 1970; 30: 2213–2222.
Heisterkamp N, Groffen J, Stephenson JR, Spurr NK, Goodfellow PN, Solomon E et al. Chromosomal localization of human cellular homologues of two viral oncogenes. Nature 1982; 299: 747–749.
Heisterkamp N, Stam K, Groffen J, de Klein A, Grosveld G . Structural organization of the bcr gene and its role in the Ph' translocation. Nature 1985; 315: 758–761.
Canaani E, Gale RP, Steiner-Saltz D, Berrebi A, Aghai E, Januszewicz E . Altered transcription of an oncogene in chronic myeloid leukaemia. Lancet 1984; 1: 593–595.
Collins SJ, Kubonishi I, Miyoshi I, Groudine MT . Altered transcription of the c-abl oncogene in K-562 and other chronic myelogenous leukemia cells. Science 1984; 225: 72–74.
Shtivelman E, Lifshitz B, Gale RP, Canaani E . Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature 1985; 315: 550–554.
Stam K, Heisterkamp N, Grosveld G, de Klein A, Verma RS, Coleman M et al. Evidence of a new chimeric bcr/c-abl mRNA in patients with chronic myelocytic leukemia and the Philadelphia chromosome. N Engl J Med 1985; 313: 1429–1433.
Ben-Neriah Y, Daley GQ, Mes-Masson AM, Witte ON, Baltimore D . The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science 1986; 233: 212–214.
Erikson J, Griffin CA, ar-Rushdi A, Valtieri M, Hoxie J, Finan J et al. Heterogeneity of chromosome 22 breakpoint in Philadelphia-positive (Ph+) acute lymphocytic leukemia. Proc Natl Acad Sci USA 1986; 83: 1807–1811.
Chan LC, Karhi KK, Rayter SI, Heisterkamp N, Eridani S, Powles R et al. A novel abl protein expressed in Philadelphia chromosome positive acute lymphoblastic leukaemia. Nature 1987; 325: 635–637.
Hermans A, Heisterkamp N, von Linden M, van Baal S, Meijer D, van der Plas D et al. Unique fusion of bcr and c-abl genes in Philadelphia chromosome positive acute lymphoblastic leukemia. Cell 1987; 51: 33–40.
Clark SS, McLaughlin J, Crist WM, Champlin R, Witte ON . Unique forms of the abl tyrosine kinase distinguish Ph1-positive CML from Ph1-positive ALL. Science 1987; 235: 85–88.
McLaughlin J, Chianese E, Witte ON . In vitro transformation of immature hematopoietic cells by the P210 BCR/ABL oncogene product of the Philadelphia chromosome. Proc Natl Acad Sci USA 1987; 84: 6558–6562.
Daley GQ, Baltimore D . Transformation of an interleukin 3-dependent hematopoietic cell line by the chronic myelogenous leukemia-specific P210bcr/abl protein. Proc Natl Acad Sci USA 1988; 85: 9312–9316.
Konopka JB, Watanabe SM, Witte ON . An alteration of the human c-abl protein in K562 leukemia cells unmasks associated tyrosine kinase activity. Cell 1984; 37: 1035–1042.
Hariharan IK, Harris AW, Crawford M, Abud H, Webb E, Cory S et al. A bcr-v-abl oncogene induces lymphomas in transgenic mice. Mol Cell Biol 1989; 9: 2798–2805.
Heisterkamp N, Jenster G, ten Hoeve J, Zovich D, Pattengale PK, Groffen J . Acute leukaemia in bcr/abl transgenic mice. Nature 1990; 344: 251–253.
Kelliher MA, McLaughlin J, Witte ON, Rosenberg N . Induction of a chronic myelogenous leukemia-like syndrome in mice with v-abl and BCR/ABL. Proc Natl Acad Sci USA 1990; 87: 6649–6653.
Daley GQ, Van Etten RA, Baltimore D . Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 1990; 247: 824–830.
Elefanty AG, Hariharan IK, Cory S . bcr–abl, the hallmark of chronic myeloid leukaemia in man, induces multiple haemopoietic neoplasms in mice. EMBO J 1990; 9: 1069–1078.
Galton DA . Myleran in chronic myeloid leukaemia; results of treatment. Lancet 1953; 264: 208–213.
Kennedy BJ, Yarbro JW . Metabolic and therapeutic effects of hydroxyurea in chronic myelogenous leukemia. Trans Assoc Am Physicians 1965; 78: 391–399.
Talpaz M, McCredie KB, Mavligit GM, Gutterman JU . Leukocyte interferon-induced myeloid cytoreduction in chronic myelogenous leukemia. Blood 1983; 62: 689–692.
Valentiner W . Ein Symptomatisch ausgezichneter Fall von Leucaemie. Berl Klin Wochenschr 1865; 2: 320–321.
Lissauer H . Zwei Falle von Leucaemie. Berl Klin Wochenschr 1865; 2: 403–404.
Senn N . Case of splenomedullary leukaemia successfully treated by the use of the Rontgen ray. Med Rec 1903; 64: 281–282.
Forkner CE, Scott TFM . Arsenic as a therapeutic agent in chronic myelogenous leukemia. JAMA 1931; 97: 3–5.
Hueper WC, Russell M . Some immunological aspects of leukemia. Arch Intern Med 1932; 49: 113–122.
Kalapos I . Die Wirkung des Benzols bei der Leukamie. Klin Wochenschr 1935; 14: 864–867.
Cooper T, Watkins CH . Ethyl carbamate (urethane) in the treatment of chronic myelocytic leukemia; results of a three-year study. Med Clin North Am 1950; 34: 1205–1215.
Osgood EE, Seaman AJ . Treatment of chronic leukemias. Results of therapy by titrated, regularly spaced total body radioactive phosphorus, or roentgen irradiation. JAMA 1952; 150: 1372–1379.
Shay H, Gruenstein M, Harris C . The effect of triethylene thiophosphoramide (thio-TEPA) in the treatment of chronic myelocytic chloroleukemia in the rat. J Natl Cancer Inst 1954; 15: 463–481.
Buckner D, Graw Jr RG, Eisel RJ, Henderson ES, Perry S . Leukapheresis by continuous flow centrifugation (CFC) in patients with chronic myelocytic leukemia (CML). Blood 1969; 33: 353–369.
Cutting HO . The effect of splenectomy in chronic granulocytic leukemia. Report of a case. Arch Intern Med 1967; 120: 356–360.
Tura S, Baccarani M, Mandelli F, Amadori S, Cajozzo A, di Marco P et al. Splenectomy in early chronic myeloid leukaemia: preliminary report on 37 cases. Haematologica 1974; 59: 428–439.
Baker MA, Taub RN, Carter Jr WH, Davidson M, Sutton DM, Kutas G et al. Immunotherapy for chronic myelogenous leukemia: survival not affected by treatment in the stable phase. Cancer Res 1984; 44: 383–385.
Cunningham I, Gee T, Dowling M, Chaganti R, Bailey R, Hopfan S et al. Results of treatment of Ph′+ chronic myelogenous leukemia with an intensive treatment regimen (L-5 protocol). Blood 1979; 53: 375–395.
Kantarjian HM, Vellekoop L, McCredie KB, Keating MJ, Hester J, Smith T et al. Intensive combination chemotherapy (ROAP 10) and splenectomy in the management of chronic myelogenous leukemia. J Clin Oncol 1985; 3: 192–200.
Chronic granulocytic leukaemia: comparison of radiotherapy and busulphan therapy. Report of the Medical Research Council's working party for therapeutic trials in leukaemia. BMJ 1968; 1: 201–208.
Frommeyer Jr WB . Comparison of cyclophosphamide (cytoxan and uracil mustard (U-8344) in chronic granulocytic leukemia. Cancer 1964; 17: 288–296.
Kaung DT, Close HP, Whittington RM, Patno ME . Comparison of busulfan and cyclophosphamide in the treatment of chronic myelocytic leukemia. Cancer 1971; 27: 608–612.
Minot GR, Buckman TE, Isaacs R . Chronic myelogenous leukemia. Age, incidence, duration, and benefit derived from radiation. JAMA 1924; 82: 1489–1494.
Goeddel DV, Yelverton E, Ullrich A, Heyneker HL, Miozzari G, Holmes W et al. Human leukocyte interferon produced by E. coli is biologically active. Nature 1980; 287: 411–416.
Talpaz M, Kantarjian HM, McCredie K, Trujillo JM, Keating MJ, Gutterman JU . Hematologic remission and cytogenetic improvement induced by recombinant human interferon alpha A in chronic myelogenous leukemia. N Engl J Med 1986; 314: 1065–1069.
Interferon alfa-2a as compared with conventional chemotherapy for the treatment of chronic myeloid leukemia. The Italian Cooperative Study Group on chronic myeloid leukemia. N Engl J Med 1994; 330: 820–825.
Hehlmann R, Heimpel H, Hasford J, Kolb HJ, Pralle H, Hossfeld DK et al. Randomized comparison of busulfan and hydroxyurea in chronic myelogenous leukemia: prolongation of survival by hydroxyurea. The German CML Study Group. Blood 1993; 82: 398–407.
Robin M, Guardiola P, Devergie A, Yeshurun M, Shapiro S, Esperou H et al. A 10-year median follow-up study after allogeneic stem cell transplantation for chronic myeloid leukemia in chronic phase from HLA-identical sibling donors. Leukemia 2005; 19: 1613–1620.
Buchdunger E, Zimmermann J, Mett H, Meyer T, Muller M, Druker BJ et al. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res 1996; 56: 100–104.
Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J . Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science 2000; 289: 1938–1942.
O'Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 2003; 348: 994–1004.
Druker BJ, Guilhot F, O'Brien SG, Gathmann I, Kantarjian H, Gattermann N et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 2006; 355: 2408–2417.
Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM et al. Activity of a specific inhibitor of the BCR–ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med 2001; 344: 1038–1042.
Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL . Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 2004; 305: 399–401.
Azam M, Nardi V, Shakespeare WC, Metcalf III CA, Bohacek RS, Wang Y et al. Activity of dual SRC-ABL inhibitors highlights the role of BCR/ABL kinase dynamics in drug resistance. Proc Natl Acad Sci USA 2006; 103: 9244–9249.
Kantarjian H, Giles F, Wunderle L, Bhalla K, O’Brien S, Wassmann B et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med 2006; 354: 2542–2551.
Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med 2006; 354: 2531–2541.
Druker BJ . Circumventing resistance to kinase-inhibitor therapy. N Engl J Med 2006; 354: 2594–2596.
Carter TA, Wodicka LM, Shah NP, Velasco AM, Fabian MA, Treiber DK et al. Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases. Proc Natl Acad Sci USA 2005; 102: 11011–11016.
Verstovsek S, Silver RT, Cross NC, Tefferi A . JAK2V617F mutational frequency in polycythemia vera: 100%, >90%, less? Leukemia 2006; 20: 2067.
Tefferi A, Lasho TL, Schwager SM, Strand JS, Elliott M, Mesa R et al. The clinical phenotype of wild-type, heterozygous, and homozygous JAK2V617F in polycythemia vera. Cancer 2006; 106: 631–635.
James C, Delhommeau F, Marzac C, Teyssandier I, Couedic JP, Giraudier S et al. Detection of JAK2 V617F as a first intention diagnostic test for erythrocytosis. Leukemia 2006; 20: 350–353.
Tefferi A, Strand JJ, Lasho TL, Knudson RA, Finke CM, Gangat N et al. Bone marrow JAK2V617F allele burden and clinical correlates in polycythemia vera. Leukemia 2007; 21: 2074–2075.
Campbell PJ, Scott LM, Buck G, Wheatley K, East CL, Marsden JT et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet 2005; 366: 1945–1953.
Wolanskyj AP, Lasho TL, Schwager SM, McClure RF, Wadleigh M, Lee SJ et al. JAK2 mutation in essential thrombocythaemia: clinical associations and long-term prognostic relevance. Br J Haematol 2005; 131: 208–213.
Kittur J, Knudson RA, Lasho TL, Finke CM, Gangat N, Wolanskyj AP et al. Clinical correlates of JAK2V617F allele burden in essential thrombocythemia. Cancer 2007; 109: 2279–2284.
Tefferi A, Lasho TL, Schwager SM, Steensma DP, Mesa RA, Li CY et al. The JAK2 tyrosine kinase mutation in myelofibrosis with myeloid metaplasia: lineage specificity and clinical correlates. Br J Haematol 2005; 131: 320–328.
Renneville A, Quesnel B, Charpentier A, Terriou L, Crinquette A, Lai JL et al. High occurrence of JAK2 V617 mutation in refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Leukemia 2006; 20: 2067–2070.
Wang SA, Hasserjian RP, Loew JM, Sechman EV, Jones D, Hao S et al. Refractory anemia with ringed sideroblasts associated with marked thrombocytosis harbors JAK2 mutation and shows overlapping myeloproliferative and myelodysplastic features. Leukemia 2006; 20: 1641–1644.
Vizmanos JL, Ormazabal C, Larrayoz MJ, Cross NC, Calasanz MJ . JAK2 V617F mutation in classic chronic myeloproliferative diseases: a report on a series of 349 patients. Leukemia 2006; 20: 534–535.
Kremer M, Horn T, Dechow T, Tzankov A, Quintanilla-Martinez L, Fend F . The JAK2 V617F mutation occurs frequently in myelodysplastic/myeloproliferative diseases, but is absent in true myelodysplastic syndromes with fibrosis. Leukemia 2006; 20: 1315–1316.
Steensma DP, McClure RF, Karp JE, Tefferi A, Lasho TL, Powell HL et al. JAK2 V617F is a rare finding in de novo acute myeloid leukemia, but STAT3 activation is common and remains unexplained. Leukemia 2006; 20: 971–978.
Nishii K, Nanbu R, Lorenzo VF, Monma F, Kato K, Ryuu H et al. Expression of the JAK2 V617F mutation is not found in de novo AML and MDS but is detected in MDS-derived leukemia of megakaryoblastic nature. Leukemia 2007; 21: 1337–1338.
Steensma DP, Dewald GW, Lasho TL, Powell HL, McClure RF, Levine RL et al. The JAK2 V617F activating tyrosine kinase mutation is an infrequent event in both ‘atypical’ myeloproliferative disorders and myelodysplastic syndromes. Blood 2005; 106: 1207–1209.
Fiorini A, Farina G, Reddiconto G, Palladino M, Rossi E, Za T et al. Screening of JAK2 V617F mutation in multiple myeloma. Leukemia 2006; 20: 1912–1913.
Melzner I, Weniger MA, Menz CK, Moller P . Absence of the JAK2 V617F activating mutation in classical Hodgkin lymphoma and primary mediastinal B-cell lymphoma. Leukemia 2006; 20: 157–158.
Tefferi A, Vardiman JW . The diagnostic interface between histology and molecular tests in myeloproliferative disorders. Curr Opin Hematol 2007; 14: 115–122.
Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 2006; 108: 3472–3476.
Lasho TL, Pardanani A, McClure RF, Mesa RA, Levine RL, Gary Gilliland D et al. Concurrent MPL515 and JAK2V617F mutations in myelofibrosis: chronology of clonal emergence and changes in mutant allele burden over time. Br J Haematol 2006; 135: 683–687.
Pardanani A, Lasho TL, Finke C, Hanson CA, Tefferi A . Prevalence and clinicopathologic correlates of JAK2 exon 12 mutations in JAK2V617F-negative polycythemia vera. Leukemia 2007; 21: 1960–1963.
Tefferi A, Gilliland DG . Oncogenes in myeloproliferative disorders. Cell Cycle 2007; 6: 550–566.
Tefferi A, Pardanani A . Evaluation of ‘increased’ hemoglobin in the JAK2 mutations era: a diagnostic algorithm based on genetic tests. Mayo Clin Proc 2007; 82: 599–604.
Tefferi A . JAK2 mutations in polycythemia vera—molecular mechanisms and clinical applications. N Engl J Med 2007; 356: 444–445.
Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med 2003; 348: 1201–1214.
Apperley JF, Gardembas M, Melo JV, Russell-Jones R, Bain BJ, Baxter EJ et al. Response to imatinib mesylate in patients with chronic myeloproliferative diseases with rearrangements of the platelet-derived growth factor receptor beta. N Engl J Med 2002; 347: 481–487.
Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002; 347: 472–480.
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001; 344: 783–792.
Smith I, Procter M, Gelber RD, Guillaume S, Feyereislova A, Dowsett M et al. 2-year follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer: a randomised controlled trial. Lancet 2007; 369: 29–36.
Coiffier B, Lepage E, Briere J, Herbrecht R, Tilly H, Bouabdallah R et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 2002; 346: 235–242.
Huang ME, Ye YC, Chen SR, Chai JR, Lu JX, Zhoa L et al. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 1988; 72: 567–572.
Tallman MS, Andersen JW, Schiffer CA, Appelbaum FR, Feusner JH, Ogden A et al. All-trans-retinoic acid in acute promyelocytic leukemia. N Engl J Med 1997; 337: 1021–1028.
Pardanani A, Hood J, Lasho T, Levine RL, Martin MB, Noronha G et al. TG101209, a small molecule JAK2-selective kinase inhibitor potently inhibits myeloproliferative disorder-associated JAK2V617F and MPLW515L//K mutations. Leukemia 2007: 21: 1658–1668.
Niblack J . http://www.mpdinfo.org/:, accessed June 15, 2007.
Rosen R . http://www.mpdfoundation.org/:, accessed June 15, 2007.
Landro L . The Growing Clout Of Online Patient Groups. The Wall Street Journal 2007; 6/13/2007: http://online.wsj.com/article/SB118168968368633094.html.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tefferi, A. The history of myeloproliferative disorders: before and after Dameshek. Leukemia 22, 3–13 (2008). https://doi.org/10.1038/sj.leu.2404946
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.leu.2404946
Keywords
This article is cited by
-
Retrospective analysis of the clinical features of 172 patients with BCR-ABL1-negative chronic myeloproliferative neoplasms
Molecular Cytogenetics (2020)
-
Current Challenges in Stem Cell Transplantation in Myelofibrosis
Current Hematologic Malignancy Reports (2015)
-
Cardiac hypertrophy associated with myeloproliferative neoplasms in JAK2V617F transgenic mice
Journal of Hematology & Oncology (2014)
-
Frequencies, clinical characteristics, and outcome of somatic CALR mutations in JAK2-unmutated essential thrombocythemia
Annals of Hematology (2014)
-
Molecular Classification of Myeloproliferative Neoplasms—Pros and Cons
Current Hematologic Malignancy Reports (2013)
