
Abstract
Studying the role of a particular gene in atherosclerosis typically requires a time-consuming and often difficult process of generating double knockouts or transgenics on ApoE−/− or LDL receptor (LDLR)−/− background. Recently, it was reported that adeno-associated-virus-8 (AAV8)-mediated overexpression of PCSK9 (AAV8-PCSK9) rapidly induced hyperlipidemia. However, using this method in C57BL6 wild-type (C57) mice, it took ~3 months to develop atherosclerosis. Our partial carotid ligation model is used to rapidly develop atherosclerosis by inducing disturbed flow in the left common carotid artery within 2 weeks in ApoE−/− or LDLR−/− mice. Here, we combined these two approaches to develop an accelerated model of atherosclerosis in C57 mice. C57 mice were injected with AAV9-PCSK9 or AAV9-luciferase (control) and high-fat diet was initiated. A week later, partial ligation was performed. Compared to the control, AAV-PCSK9 led to elevated serum PCSK9, hypercholesterolemia, and rapid atherosclerosis development within 3 weeks as determined by gross plaque imaging, and staining with Oil-Red-O, Movat’s pentachrome, and CD45 antibody. These plaque lesions were comparable to the atherosclerotic lesions that have been previously observed in ApoE−/− or LDLR−/− mice that were subjected to partial carotid ligation and high-fat diet. Next, we tested whether our method can be utilized to rapidly determine the role of a particular gene in atherosclerosis. Using eNOS−/− and NOX1−/y mice on C57 background, we found that the eNOS−/− mice developed more advanced lesions, while the NOX1−/y mice developed less atherosclerotic lesions as compared to the C57 controls. These results are consistent with the previous findings using double knockouts (eNOS−/−_ApoE−/− and NOX1−/y_ApoE−/−). AAV9-PCSK9 injection followed by partial carotid ligation is an effective and time-saving approach to rapidly induce atherosclerosis. This accelerated model is well-suited to quickly determine the role of gene(s) interest without generating double or triple knockouts.
Similar content being viewed by others
Main
Atherosclerosis is a multifactorial disease that is one of the leading cause of death worldwide.1 Although multiple systemic factors such as hypercholesterolemia, diabetes, hypertension, and smoking are well-known risk factors; atherosclerosis occurs preferentially in the branched or curved arterial regions exposed to disturbed blood flow (d-flow),2 while the straight arterial regions exposed to high and stable shear stress remain protected from atherosclerosis.3 Previously, we developed a mouse model of d-flow-induced atherosclerosis by partially ligating the left carotid artery (LCA) of ApoE−/− mouse, directly demonstrating the role of d-flow in atherosclerosis.4, 5 In this model, partial carotid ligation causes induction of d-flow in the LCA that rapidly leads to development of atherosclerosis within 2 weeks following partial ligation, while the contralateral, undisturbed right common carotid artery (RCA) remains healthy and plaque-free. This model has been used as accelerated model of atherosclerosis development, but it still requires the use of ApoE−/− or LDL receptor (LDLR)−/− animals as C57BL6 wild type (C57) fails to develop noticeable atherosclerosis.4, 5, 6
To determine the mechanisms of atherosclerosis, one of the most widely used approaches is to use genetically modified animal models such as the ApoE−/− or LDLR−/− mice.7, 8, 9, 10, 11, 12, 13 In addition, to study the role of a ‘gene of interest’ in atherosclerosis, it requires the generation of double knockouts or transgenics by cross-breeding a genetically engineered mouse line to ApoE−/− or LDLR−/− background. Generating these mice is one of the most challenging, time-consuming, and costly steps. Often times, requirement of specific cell type-targeted overexpression or deletion of a gene and/or requirement of inducible expression using Cre-LoxP or similar system requires generating triple knockouts. These additional steps further increase the difficulties and result in subsequent delays in performing atherosclerosis studies. Recently, an alternative method, using a recombinant adeno-associated-virus (AAV) encoding PCSK9, was introduced that can result in atherosclerosis development without using ApoE−/− or LDLR−/− mice.14
PCSK9 is involved in cholesterol metabolism and atherosclerosis development, and its inhibitors are now used as new, cholesterol-lowering drugs for patients.15, 16, 17, 18, 19, 20, 21, 22, 23, 24 PCSK9 reduces hepatic uptake of LDL by increasing the lysosomal degradation of LDLRs, thereby generating an LDLR−/−-like phenotype.25 Recent studies showed that mice deficient for PCSK9 protein have low plasma LDL cholesterol levels and are protected against atherosclerosis development.26, 27, 28 In contrast, transgenic mice overexpressing gain-of-function mutants of PCSK9 developed hypercholesterolemia and atherosclerosis.25, 29, 30, 31 More recently, Bjorklund et al14 developed the recombinant AAV8 expressing the gain-of-function mutants of PCSK9 (AAV8-PCSK9). A single injection of AAV8-PCSK9 into C57 mice resulted in significant hypercholesterolemia and subsequent atherosclerosis development within 3 months, introducing a convenient alternative to inducing hypercholesterolemia and atherosclerosis without the need of germ line knockout of ApoE or LDLR.14
Here, we combined our partial carotid ligation model and AAV-PCSK9 method to develop an accelerated model of atherosclerosis in a flow- and hypercholesterolemia-dependent manner using C57 mice. Using this method, we show that atherosclerosis rapidly develops in the LCA within 3 weeks following partial carotid ligation and AAV-PCSK9 injection. We further demonstrate the proof-of-principle using eNOS−/− and NOX1−/y on C57 background studying the role of each gene in atherosclerosis development can be achieved without generating the double knockouts using ApoE−/− or LDLR−/− mice.
MATERIALS AND METHODS
AAV Vector Production and Purification
The gain-of-function murine PCSK9 mutant plasmid (pAAV/D377Y-mPCSK9) was a gift from Jacob Bentzon (Addgene plasmid # 58376).14 Recombinant AAV serotype-9 expressing the PCSK9 mutant under the hepatic control region-apolipoprotein enhancer/alpha1-antitrypsin, a liver-specific promoter (AAV9-HCRApoE/hAAT-D377Y-mPCSK9) was produced by the Emory Viral Vector Core at Emory University. Briefly, AAV plasmids were cloned and propagated in the DH5α Escherichia coli strain (Life Technologies). Shuttle plasmid pAAV-D377Y-mPCSK9 was packaged into capsids AAV9, using helper plasmids p-helper (providing the three adenoviral helper genes) and plasmid pAAV2/9 (providing rep and cap viral genes). The amplified AAV shuttle and helper plasmids were co-transfected into HEK 293T cells by polyethylenimine (PEI). A total of 420 μg plasmid DNA (mixed in an equimolar ratio) was used for 20 × 150 mm plates (Corning) seeded with 1 × 107 cells per plate the day before. Seventy-two hours after transfection, cell culture media and transfected cells were collected separately. Forty percent polyethylene glycol (PEG) in 2.5 N NaCl was added to the supernatant to a final concentration of 8%, and incubated on ice for 2 h. The cell pellet was suspended in 14 ml of lysis buffer (50 mM Tris-Cl, 150 mM NaCl and 2 mM MgCl2) and stored at 4 °C. Following the 2 h incubation, the supernatant was centrifuged at 2500 g for 30 min at 4 °C to pellet the PEG-precipitated virus. The cell lysate and pelleted supernatant precipitate were combined, and then treated with 750 μl of 10% sodium deoxycholate and benzonase, and three freeze-thaw cycles between −80 and 37 °C. Cell debris was pelleted by spinning at 12 000 g for 30 min at 4 °C. The lysate was purified by iodixanol gradient centrifuge followed by using an Amicon 15 100 000 MWCO concentration unit. The virus was aliquoted and stored at −80 °C. The vector genome copies (VG) were determined by qPCR using Brilliant III Ultra-Fast SYBRgreen qPCR Master Mix. The viral DNA was extracted from 1 μl of purified virus and was treated with 0.5 U DNase I to digest any contaminating unpackaged DNA, followed by an additional 10 μg proteinase K treatment to assist in breaking capsids and releasing viral DNA. qPCR was run in Applied Biosystems Mx3000P with primers for the ITRs common to AAV transfer vector plasmids: forward primer 5′-GGAACCCCTAGTGATGGAGTT-3′ and reverse primer 5′-CGGCCTCAGTGAGCGA-3′; set with a program: 95 °C 10 min, then cycled 40 times at 95 °C for 15 s, 60 °C for 30 s, and 72 °C for 30 s. To generate a standard curve, a rAAV-GFP plasmid was used in serial dilutions from 1 × 107 to 1 × 103 genome copies, performed in triplicate. In addition, high-titer recombinant AAV8 expressing the PCSK9 mutant and recombinant AAV9 expressing firefly luciferase, respectively, under the liver-specific promoter (rAAV8-D377Y-mPCSK9 and rAAV9-HCRApoE/hAAT-Luc) were purchased from Vector BioLabs (Malvern, PA).
Mouse Studies
Mice were maintained and cared for in accordance to the National Institutes of Health (NIH) guidelines in our AAALAC-accredited experimental animal facility under controlled environment (21±2 °C, 50±10% relative humidity, and a 12 h light:12 h dark cycle with lights on at 0700 h EST). All mouse studies performed here were approved by the Institutional Animal Care and Use Committee at Emory University, and were in accordance with the established guidelines and regulations consistent with federal assurance. C57BL/6J mice (cat #000664), ApoE null (B6.129P2-Apoetm1Unc/J; cat #002052), eNOS KO (B6.129P2-Nos3tm1Unc/J; cat #002684), and NOX1 knockout (Nox1tm1Kkr; cat #018787) were purchased from Jackson Laboratory (Bar Harbor, ME, USA), and housed at the Emory University animal facility. These mice were fed ad libitum with standard chow diet until a week before the surgery at 10 weeks of age.
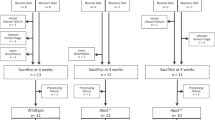
Mice were injected once with AAV-PCSK9 (1 × 1011 VG), AAV-Luc (1 × 1011 VG), or saline via tail vein (Figure 1), and fed a high-fat diet (16% fat and 1.25% cholesterol, Research Diets, cat # D12336, New Brunswick, NJ, USA). A week after AAV-PCSK9 injection, mice were subjected to partial carotid ligation surgery as we previously described4, 5 and high-fat diet was continued for another 3 weeks (Figure 1). Finally, ultrasonography was performed to determine the luminal flow and the animals were killed. Blood was collected from inferior vena cava using a heparinized syringe fitted with a 25 G needle. In some control studies, C57 mice were injected with AAV-PCSK9 or control (AAV-Luc), and fed a high fat for 3 months without the partial carotid ligation surgery. Aortic arch and carotids were collected, and gross macroscopic images were acquired as we previously reported.32, 33, 34 For molecular histology studies, the carotids were fixed with formalin and aortic roots, and arches were embedded in OCT compound and stored at −80 °C until use.

Overall study design and work flow. Mice were injected with AAV9-PCSK9 (1 × 1011 VG per mouse) once via tail vein injection and fed a high-fat diet for the entire duration of the study. Partial carotid ligation was performed 1 week after the injection. Three weeks post ligation, animals were killed, carotid arteries were explanted, and atherosclerotic plaques were quantified.
In vivo Analysis of Bioluminescence
For the bioluminescence study, C57 mice (n=5) were injected intravenously through the tail vein with AAV-Luc (1.0 × 1011 VG per mouse) and saline was used as control (n=3). Bioluminescence was analyzed 7 days later using the In-Vivo Xtreme imaging station (Bruker). Mice were anesthetized with 3.75% isoflurane and then injected intraperitoneally with luciferin (150 mg/kg body weight, Caliper LifeSciences).35 Isoflurane anesthesia was maintained at 1.5% while bioluminescence was analyzed. Images were acquired 10 min after luciferin injection. X-ray image was used for determining the anatomic landmarks.
Blood Measurements
Plasma concentrations of PCSK9 were determined using ELISA Kits from R&D systems (Minneapolis, MN, USA). Plasma total cholesterol, high-density lipoproteins, low-density lipoproteins (LDL) and very-low-density lipoproteins, and triglyceride levels were assessed by Emory Biochemistry and Pathology Laboratory as described previously.34, 36
Histochemistry and Image Quantification
Tissue samples were frozen in OCT compound and 8 μm serial sections were prepared. Lipids were detected with Oil-Red-O staining following the standard protocol as described.4 Russell Movat Pentachrome Stain Kit was used to perform pentachrome staining on serial sections (American MasterTech Scientific) as per manufacturer’s protocol.37, 38 Color images of each tissue section were acquired using Hamamatsu’s NanoZoomer Digital Pathology System (× 20 objective, NA 0.75). Plaque content included the changes in fibrin/fibrinoid tissue, mucin, ground substance, and collagen staining, and was quantified using the NIH Image-J software.39
Immunofluorescence Staining
Tissue samples were cut into 8 μm thin slices of histological sections, and cryosections were fixed in acetone. After blocking in 4% serum, sections were incubated with fluorescently labeled primary antibody (CD45). After labeling with appropriate secondary antibody, sections were washed in PBS and embedded in a mounting medium containing DAPI (Vector Laboratories). Slides were examined using a confocal microscope (Zeiss, Germany). Images were analyzed and quantification was performed using the NIH Image-J software.39
Preparation of Liver Homogenates and Western Blotting
Liver homogenates were prepared in RIPA buffer (Santa Cruz Biotechnology) containing protease and phosphatase inhibitors. Briefly, a portion of mouse liver tissue was homogenized in an ice cold 1.5 ml tube with a disposable plastic pestle using ice cold RIPA buffer containing freshly dissolved protease and phosphatase inhibitor pellets. Samples were run through a syringe fitted with 22 gauge needle and then centrifuged at 12 000 g for 15 min at 4 °C. Supernatants were collected and total protein was quantified using the BCA assay (Thermo Scientific), and 50 μg of protein in SDS sample loading buffer (Boston Bioproducts, Ashland, MA, USA) was loaded onto a 10% SDS-PAGE gel. Gels were transferred onto PVDF membranes, blocked in 5% milk for 45 min at room temperature, and incubated overnight at 4 °C in primary antibody (1:1000 LDLR antibody, BioVision). Post washing, appropriate secondary antibodies (1:5000 anti-rabbit; Bio-Rad) were used to develop and photographically capture the luminescent signal on an X-ray film using an enhanced chemiluminescent substrate (Thermo Fisher).
Quantification and Morphology of Atherosclerotic Lesions
Lesion development in the whole aortic tree was determined using the en face Oil-Red-O staining technique. Briefly, the entire aorta was removed and cleaned for periadventitial fat, cut open longitudinally, and fixed on black paraffin wax dish using insect mounting pins. These opened up aortic trees were then stained with Oil-Red-O and images were taken using stereomicroscope equipped with a camera, as described previously.34 The percentage of total area stained by Oil-Red-O was determined using the NIH Image-J software,39 using methodology as previously described.33, 40 Lesion morphology and atherosclerosis development in the carotid arteries were studied initially by gross imaging of the carotid arteries followed by performing Oil-Red-O staining on the cross-sections of the RCAs and LCAs.
Statistical Analysis
Data are shown as mean±s.e.m.; n indicates the number of mice. Statistical analyses were performed using GraphPad Prism Version 7 (Prism Software, La Jolla, CA, USA). For comparison between two groups, a paired or unpaired two-tailed Student’s t-test with equal or unequal variances was performed. For comparison among three or more treatment groups, one-way ANOVA followed by Bonferroni’s post hoc test was carried out. The quantitative analyses for histology were performed by two investigators blinded to the treatment groups. A P≤0.05 was considered statistically significant.
RESULTS
AAV-PCSK9 Effectively Reduces LDLRs in Mouse
Our goal was to express PCSK9 in the mouse liver to effectively knock down the LDLRs. To this end, we used AAV9 and the liver-specific HCRApoE/hAAT promoter to construct the recombinant AAV9-PCSK9 and AAV9-Luc as an expression control. To determine the liver-specific expression of our AAV9 constructs, we first studied the expression of AAV-Luc by in vivo bioluminescence assay. As shown in Figures 2a and b, intense luciferase expression was detected in the liver injected with AAV-Luc, but not in the saline treated control group. More importantly, we found that a single AAV9-PCSK9 injection reduced LDLR expression by >90% in the liver compared to the AAV9-Luc control group (Figures 2c and d). In addition, AAV-PCSK9 injection and high-fat diet induced a significant hypercholesterolemia (total cholesterol ~700 mg/dl) from 1 week and up to 3 months (Supplementary Table 1). These results show that AAV9-mediated PCSK9 delivery is an effective method to knock down LDLRs and induce hypercholesterolemia.

AAV-PCSK9 knocks down LDL receptor level in liver in C57 mice. (a, b) In vivo imaging of luciferase activity after tail vein injection of AAV-Luc. Mice were injected with AAV9-Luc (n=5) or saline (n=3) via tail vein, and after 7 days, the luciferase activity was imaged using in vivo imager. X-ray image was also taken and superimposed on bioluminescence images to identify the anatomical landmarks. (c, d) Western blot analysis showing the expression level of LDL receptors in the liver tissue samples obtained from animals injected with AAV-PCSK9 or AAV-Luc. Western blot results were quantified using Image-J software. Means±s.e.m., *P<0.05, n=5.
AAV9-PCSK9 Injection Induces Atherosclerosis in C57 mice Fed High-Fat Diet within 3 Months
We next tested whether AAV-PCSK9 induces atherosclerosis in C57 mice fed a high-fat diet for 3 months. As expected and consistent with the previous reports, we found that AAV9-PCSK9 induced robust atherosclerotic plaques in the aortic arch as shown by the gross imaging (Figures 3a and b) and in the whole aortic tree as shown by the en face Oil-Red-O staining (Figures 3c and d). Plaque lesion area in the lesser curvature (LC), greater curvature, and thoracic aorta was quantified using Image-J (Supplementary Figure 1). In contrast, mice injected with the AAV9-Luc (control) did not develop any significant atherosclerotic plaques (Figures 3a–d). These results confirm that AAV-PCSK9 and high-fat diet is an effective way to induce atherosclerosis in C57 mice. We also observed a significant increase in the levels of total cholesterol, triglycerides, and LDL in the plasma samples obtained from AAV-PCSK9-treated group compared to the AAV9-Luc (control) group (Figure 3e).

AAV-PCSK9 induces hypercholesterolemia and atherosclerosis within 3 months in C57 mice. (a) C57 mice were injected with AAV-PCSK9 or AAV-Luc (1 × 1011 VG/mouse), and fed a high-fat diet for 3 months. Atherosclerotic plaques in the aortic arch were imaged and quantified (a, c). Means±s.e.m., *P<0.05, n=5. Also, the arterial trees were dissected out, and en face Oil-Red-O stained and quantified (b, d). Means±s.e.m., *P<0.05, n=5. (e) Plasma triglycerides, total cholesterol, HDL, and LDL (mg/dl) from mice treated with AAV-PCSK9 or AAV-Luc at 3 months.
Partial Carotid Ligation Further Accelerates Atherosclerosis Development in AAV9-PCSK9-Treated C57 Mice
Partial carotid ligation rapidly induces atherosclerosis by causing d-flow in the LCA within 2 weeks in ApoE−/− mice fed a high-fat diet. In contrast, high-fat diet alone without the partial carotid ligation surgery takes 3 or more months to develop significant atherosclerotic plaques either in ApoE−/− or AAV-PCSK9-injected mice. Here, we tested whether we can reduce the time it takes to develop significant atherosclerotic plaques by combining the partial carotid ligation surgery and AAV-PCSK9 injection in C57 mice. As shown in Figure 4a, partial carotid ligation surgery plus high-fat diet induced robust atherosclerosis in the LCA within 3 weeks in mice injected with AAV-PCSK9 but not in the AAV-Luc control group (Figures 4a and d). As an additional built-in control in each animal, the contralateral non-ligated RCA (exposed to stable flow) remained virtually plaque-free. The Oil-Red-O staining using the frozen sections prepared from the carotid arteries showed a significant increase in the plaque lesion area in the LCAs of AAV-PCSK9-treated group compared to the control group (Figures 4g and j). These results demonstrate that partial carotid ligation significantly reduces the time it takes to develop significant atherosclerosis in a flow- and hypercholesterolemia-dependent manner.


Partial carotid ligation of C57 mice treated with AAV-PCSK9 and high-fat diet induces atherosclerosis within 3 weeks—the roles of eNOS−/− and NOX1−/y on C57 background. Mice were injected with AAV-PCSK9 or AAV-Luc (control) (1 × 1011 VG per mouse) and fed a high-fat diet for the entire duration of the study. After 1 week, partial carotid ligation was performed and animals were killed at 3 weeks post surgery. RCAs and LCAs were dissected out and atherosclerotic plaques were imaged using dissection microscopy in (a) C57 (b) eNOS−/−, and (c) NOX1−/y mice, respectively, and quantified by Image-J. Means±s.e.m., *P<0.05, n=5 (d–f). Red arrows shown in a–c depict the region from which the cross-sections of the carotid arteries were obtained and quantified in g–l. (g–i) Oil-Red-O staining of the serial sections obtained from C57, eNOS−/−, and NOX1−/y mice, respectively are shown. Inset shows zoomed-in images. Quantification of Oil-Red-O positive staining (j–l). Means±s.e.m., *P<0.05, n=5.
Roles of eNOS and NOX1 in Atherosclerosis can be Easily Tested in C57 Mice by Combining Partial Carotid Ligation and AAV-PCSK9
Previously, it required generation of double knockout or transgenic mice to determine the role of gene(s) of interest in atherosclerosis. For example, previous studies had to generate double knockout mice by crossing eNOS−/− or NOX1−/y with ApoE−/− mice. These studies showed that eNOS−/− mice on ApoE−/− background show exacerbated atherosclerosis development,41, 42, 43 while the NOX1−/y mice on ApoE−/− resists atherosclerosis development.44, 45 As a proof-of-principle, here we tested whether a combined approach viz. partial carotid ligation and AAV-PCSK9 injection, can be used to study the role of eNOS−/− or NOX1−/y in atherosclerosis without using the respective double knockouts mice. To this end, we injected AAV9-PCSK9 in eNOS−/− and NOX1−/y mice on C57 background, and performed partial carotid ligation and fed high-fat diet. We found that eNOS−/− mice developed robust plaques in the LCA within 3 weeks (Figure 4b), compared to the controls (Figure 4a), while the plaque development in the NOX1−/y animals was significantly reduced compared to the C57 controls (Figures 4c and a). As expected, the contralateral non-ligated RCAs remained virtually plaque-free in all the experimental groups. In all the three groups of mice, AAV-PCSK9 injection resulted in similar levels of hypercholesterolemia and serum PCSK9 (Supplementary Figures 2 and 3). Supplementary Figure 4 illustrates additional gross plaque images showing atherosclerosis development in C57, eNOS−/−, and NOX1−/y mice using AAV-PCSK9, high-fat diet, and partial carotid ligation surgery.
To further characterize the details of plaque phenotypes, we performed Movat’s pentachrome staining, and compared the samples from C57, eNOS−/−, and NOX1−/y mice treated with AAV-PCSK9 and partial carotid ligation for 3 weeks. We observed a significant increase in the fibrin/fibrinoid tissues in the LCA of eNOS−/− mice, compared to the C57 controls (Figures 5a, b, d and e). Interestingly, the fibrin/fibrinoid tissues, collagen deposition in the LCAs of NOX1−/y mice were significantly less compared to the C57 and eNOS−/− mice (Figure 4). These results are consistent with the previous findings using eNOS−/−_ApoE−/− and NOX1−/y_ApoE−/− double knockout mice.42, 43, 44, 46

Effect of eNOS−/− and NOX1−/y on atherosclerotic plaque phenotype in mice subjected to partial carotid ligation and AAV-PCSK9 treatment on C57 background. Serial sections obtained (as described above in Figure 4) were subjected by Movat’s pentachrome staining; (a) C57, (b) eNOS−/−, and (c) NOX1−/y mice, respectively, and the inset shows zoomed-in images (scale bar=100 μm). Image-J quantification of relative fibrin/fibrinoid staining (red), ground substance/mucin (blue), and collagen (yellow) are shown (d–f). The intensity of respective staining in the RCA of control animal was arbitrarily set at 1; means±s.e.m., *P<0.05, n=5.
In addition, CD45 immunostaining showed that macrophage accumulation in the LCAs was significantly increased in eNOS−/− mice compared to the C57 controls, while the LCAs of NOX1−/y mice showed significantly reduced macrophage accumulation (Figures 6a–c). The contralateral non-ligated RCAs of AAV9-PCSK9 injected group as well as the controls remained free of macrophage accumulation (Figures 6a–g). As a comparison, CD45 immunostaining on the ApoE−/− animals subjected to partial carotid ligation surgery and high-fat feeding for same duration showed comparable macrophage accumulation in their LCAs (Figure 6d). Together, these results demonstrate that the combined partial carotid ligation and AAV-PCSK9 method can be used to rapidly test the role of genes in atherosclerosis in flow- and cholesterol-dependent manner in C57 mice without generating the double knockouts on ApoE−/− or LDLR−/− background.

Effect of eNOS−/− and NOX1−/y on atherosclerotic plaque macrophage content in mice subjected to partial carotid ligation and AAV-PCSK9 treatment on C57 background. Serial sections obtained (as described above in Figure 4) were subjected to immunofluorescence staining using anti-CD45 antibody; (a) C57, (b) eNOS−/−, and (c) NOX1−/y mice, respectively, and the inset shows zoomed-in images (scale bar=100 μm). (d) Immunostaining for CD45.1 using the RCA and LCA cross-sections from ApoE−/− mice subjected to partial carotid ligation and fed a high-fat diet for 3 weeks were used a positive control. (e–g) Image-J was used to quantify CD45.1 staining intensity in the RCAs and LCAs of C57, eNOS−/− mice and NOX1−/y, respectively. For comparison, quantification of CD45.1 staining from the RCAs and LCAs of ApoE−/− mice subjected to partial carotid ligation is also plotted in e. Means±s.e.m.; *P<0.05, n=5. DAPI was used for counterstaining nuclei (blue). Autofluorescence (green) shows internal elastic lamina. White scale bar=100 μm.
DISCUSSION
Our results show that a single injection of AAV9-PCSK9, in conjunction with partial carotid ligation surgery and a high-fat diet rapidly induces robust atherosclerosis in the flow-disturbed LCA in C57 mice within 3 weeks. Using this new methodology in two independent knockout mouse lines (eNOS−/− and NOX1−/y on C57 background), we further demonstrated that the role of these genes in atherosclerosis can be studied without the need to generate the double knockouts on ApoE−/− or LDLR−/− background. These results suggest that virtually any gene(s) can be rapidly studied for its role in atherosclerosis without having to generate double knockouts on ApoE−/− or LDLR−/− background and wait months for atherosclerosis development.
Recent studies have reported that a single AAV-PCSK9 injection in C57 wild-type mice can induce hypercholesterolemia and atherosclerosis,14, 29 but it took 12–16 weeks to develop atherosclerosis in these animals. More recently, the same methodology has been used to induce abdominal aortic aneurysms and vascular calcification in experimental mouse models.30, 47 Here, we combined the AAV-PCSK9 method with our partial carotid ligation model to study the contribution of d-flow to atherosclerosis development. We and others have previously shown that induction of d-flow by partial carotid ligation induces endothelial dysfunction and accelerates atherosclerosis using ApoE−/− or LDLR−/− mice.4, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58 Another important advantage of AAV-mediated expression of PCSK9 is that it is simple, and provides a rapid (~1 week after the injection) and sustained (>1 year) elevation of blood PCSK9 and cholesterol levels by a single injection.14, 29
Our partial carotid ligation provides an additional advantage of having a built-in control of the RCA (RCA exposed to stable flow protecting against atherosclerosis) in comparison to the LCA (exposed to d-flow leading to atherosclerosis) in each mouse. This makes it easier to determine the role of each gene in atherosclerosis in a flow and hypercholesterolemic manner in animal models in a much reduced time without having to wait for a few months for plaques to develop. The main reason that our partial carotid ligation model develops atherosclerosis faster than other conventional models (for example, high-fat diet only model) is due to the exaggerated and sustained flow disturbance in the flow-modified carotid (LCA). Although, atherosclerosis is a multifactorial disease and plaque development requires additional factors (such as hypercholesterolemia), disturbed flow regions are where atherosclerosis begins to develop. It is important to note that the molecular mechanisms underlying the partial carotid ligation-induced atherosclerosis are identical to that observed in the naturally athero-prone areas, such as the LC of the aortic arch. We previously showed that many of the flow-sensitive genes and microRNAs (VCAM1, DNMT1, miR-712, and miR-205) upregulated in the naturally and endogenously disturbed flow regions, such as aortic arch are also upregulated in our partial carotid ligation model.34, 54 Our findings suggest that the mechanistic insights obtained from the partial carotid ligation model applies well to other models of atherosclerosis.
Interestingly, our results show that eNOS deficiency leads to acceleration of atherosclerosis under disturbed flow conditions, but stable flow-exposed regions remained protected. This suggests that the atheroprotective effect of stable flow is mediated by other factors that could compensate for eNOS deficiency in the RCA. These additional compensatory factors under stable flow conditions may target important anti-atherogenic genes in endothelium that may play a role in inhibition of leukocyte adhesion, and/or protection of the endothelial permeability barrier.59, 60, 61, 62
In this study, we used rAAV9 serotype, although previous studies have used the rAAV8 serotype to deliver the gain-of-function mutant of PCSK9.14 The reason to use rAAV9 was based on the previous finding by Gao et al,63 which showed its superior transduction in the liver as compared to AAV8. However, we found that there was no significant difference in the development of atherosclerosis between these serotypes (data not shown).
In conclusion, we show that the combination of a single AAV-PCSK9 injection, high-fat diet, and partial carotid ligation rapidly induces robust atherosclerosis in the flow-disturbed carotid artery within 3 weeks in C57 mice. Our results suggest that this is a quick and convenient model to study atherosclerosis and mechanisms using any knockout or transgenic mice without having to generate double knockouts on ApoE−/− or LDLR−/− background. Our combined in vivo model is well-suited for testing various therapeutic interventions targeting atherosclerosis in a considerably reduced study duration compared to the conventional high-fat diet only model.
References
Weber C, Noels H . Atherosclerosis: current pathogenesis and therapeutic options. Nat Med 2011;17:1410–1422.
Tarbell JM, Shi ZD, Dunn J et al, Fluid mechanics, arterial disease, and gene expression. Annu Rev Fluid Mech 2014;46:591–614.
Kumar S, Kim CW, Son DJ et al, Flow-dependent regulation of genome-wide mRNA and microRNA expression in endothelial cells in vivo. Sci Data 2014;1:140039.
Nam D, Ni CW, Rezvan A et al, Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. Am J Physiol Heart Circ Physiol 2009;297:H1535–H1543.
Nam D, Ni CW, Rezvan A et al, A model of disturbed flow-induced atherosclerosis in mouse carotid artery by partial ligation and a simple method of RNA isolation from carotid endothelium. J Vis Exp 2010;40:e1861.
Ni CW, Qiu H, Rezvan A et al, Discovery of novel mechanosensitive genes in vivo using mouse carotid artery endothelium exposed to disturbed flow. Blood 2010;116:e66–e73.
Shimada M, Ishibashi S, Inaba T et al, Suppression of diet-induced atherosclerosis in low density lipoprotein receptor knockout mice overexpressing lipoprotein lipase. Proc Natl Acad Sci USA 1996;93:7242–7246.
Ishibashi S, Goldstein JL, Brown MS et al, Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice. J Clin Invest 1994;93:1885–1893.
Herijgers N, Van Eck M, Groot PH et al, Effect of bone marrow transplantation on lipoprotein metabolism and atherosclerosis in LDL receptor-knockout mice. Arterioscler Thromb Vasc Biol 1997;17:1995–2003.
Tomita H, Hagaman J, Friedman MH et al, Relationship between hemodynamics and atherosclerosis in aortic arches of apolipoprotein E-null mice on 129S6/SvEvTac and C57BL/6 J genetic backgrounds. Atherosclerosis 2012;220:78–85.
Smith DD, Tan X, Tawfik O et al, Increased aortic atherosclerotic plaque development in female apolipoprotein E-null mice is associated with elevated thromboxane A2 and decreased prostacyclin production. J Physiol Pharmacol 2010;61:309–316.
Wang YX, Martin-McNulty B, Huw LY et al, Anti-atherosclerotic effect of simvastatin depends on the presence of apolipoprotein E. Atherosclerosis 2002;162:23–31.
Lutgens E, Faber B, Schapira K et al, Gene profiling in atherosclerosis reveals a key role for small inducible cytokines: validation using a novel monocyte chemoattractant protein monoclonal antibody. Circulation 2005;111:3443–3452.
Bjorklund MM, Hollensen AK, Hagensen MK et al, Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circ Res 2014;114:1684–1689.
Cainzos-Achirica M, Martin SS, Cornell JE et al, PCSK9 inhibitors: a new era in lipid-lowering treatment? Ann Intern Med 2015;163:64–65.
Gencer B, Rodondi N, Mach F . PCSK9 inhibitors: emerging treatment to lower cholesterol? Rev Med Suisse 2014;10:544.
Langslet G . Replacing statins with PCSK9-inhibitors and delaying treatment until 18 years of age in patients with familial hypercholesterolaemia is not a good idea. Eur Heart J 2016;37:1357–1359.
Saussele T . PCSK9 inhibitors. A new approach for treatment of hypercholesterolemia. Med Monatsschr Pharm 2015;38:250–257.
Tice JA, Kazi DS, Pearson SD . Proprotein convertase. subtilisin/kexin type 9 (PCSK9) inhibitors for treatment of high cholesterol levels: effectiveness and value. JAMA Intern Med 2016;176:107–108.
Marian AJ . PCSK9 as a therapeutic target in atherosclerosis. Curr Atheroscler Rep 2010;12:151–154.
Li S, Li JJ . PCSK9: a key factor modulating atherosclerosis. J Atheroscler Thromb 2015;22:221–230.
Li S, Guo YL, Xu RX et al, Plasma PCSK9 levels are associated with the severity of coronary stenosis in patients with atherosclerosis. Int J Cardiol 2014;174:863–864.
Giunzioni I, Tavori H . New developments in atherosclerosis: clinical potential of PCSK9 inhibition. Vasc Health Risk Manag 2015;11:493–501.
Abboud S, Karhunen PJ, Lutjohann D et al, Proprotein convertase subtilisin/kexin type 9 (PCSK9) gene is a risk factor of large-vessel atherosclerosis stroke. PLoS ONE 2007;2:e1043.
Somanathan S, Jacobs F, Wang Q et al, AAV vectors expressing LDLR gain-of-function variants demonstrate increased efficacy in mouse models of familial hypercholesterolemia. Circ Res 2014;115:591–599.
Le May C, Kourimate S, Langhi C et al, Proprotein convertase subtilisin kexin type 9 null mice are protected from postprandial triglyceridemia. Arterioscler Thromb Vasc Biol 2009;29:684–690.
Creemers JW, Khatib AM . Knock-out mouse models of proprotein convertases: unique functions or redundancy? Front Biosci 2008;13:4960–4971.
Ai D, Chen C, Han S et al, Regulation of hepatic LDL receptors by mTORC1 and PCSK9 in mice. J Clin Invest 2012;122:1262–1270.
Roche-Molina M, Sanz-Rosa D, Cruz FM et al, Induction of sustained hypercholesterolemia by single adeno-associated virus-mediated gene transfer of mutant hPCSK9. Arterioscler Thromb Vasc Biol 2015;35:50–59.
Goettsch C, Hutcheson JD, Hagita S et al, A single injection of gain-of-function mutant PCSK9 adeno-associated virus vector induces cardiovascular calcification in mice with no genetic modification. Atherosclerosis 2016;251:109–118.
Tavori H, Giunzioni I, Predazzi IM et al, Human PCSK9 promotes hepatic lipogenesis and atherosclerosis development via apoE- and LDLR-mediated mechanisms. Cardiovasc Res 2016;110:268–278.
Go YM, Son DJ, Park H et al, Disturbed flow enhances inflammatory signaling and atherogenesis by increasing thioredoxin-1 level in endothelial cell nuclei. PLoS ONE 2014;9:e108346.
Kim CW, Song H, Kumar S et al, Anti-inflammatory and antiatherogenic role of BMP receptor II in endothelial cells. Arterioscler Thromb Vasc Biol 2013;33:1350–1359.
Son DJ, Kumar S, Takabe W et al, The atypical mechanosensitive microRNA-712 derived from pre-ribosomal RNA induces endothelial inflammation and atherosclerosis. Nat Commun 2013;4:3000.
Zincarelli C, Soltys S, Rengo G et al, Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther 2008;16:1073–1080.
Kheirolomoom A, Kim CW, Seo JW et al, Multifunctional nanoparticles facilitate molecular targeting and miRNA delivery to inhibit atherosclerosis in ApoE(−/−) mice. ACS Nano 2015;9:8885–8897.
Russell HK Jr . A modification of Movat's pentachrome stain. Arch Pathol 1972;94:187–191.
Movat HZ . Demonstration of all connective tissue elements in a single section; pentachrome stains. AMA Arch Pathol 1955;60:289–295.
Schneider CA, Rasband WS, Eliceiri KW . NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012;9:671–675.
Andres-Manzano MJ, Andres V, Dorado B . Oil Red O and hematoxylin and eosin staining for quantification of atherosclerosis burden in mouse aorta and aortic root. Methods Mol Biol 2015;1339:85–99.
Knowles JW, Reddick RL, Jennette JC et al, Enhanced atherosclerosis and kidney dysfunction in eNOS(–/–)Apoe(–/–) mice are ameliorated by enalapril treatment. J Clin Invest 2000;105:451–458.
Kuhlencordt PJ, Gyurko R, Han F et al, Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation 2001;104:448–454.
Ponnuswamy P, Schrottle A, Ostermeier E et al, eNOS protects from atherosclerosis despite relevant superoxide production by the enzyme in apoE mice. PLoS ONE 2012;7:e30193.
Sheehan AL, Carrell S, Johnson B et al, Role for Nox1 NADPH oxidase in atherosclerosis. Atherosclerosis 2011;216:321–326.
Gray SP, Di Marco E, Okabe J et al, NADPH oxidase 1 plays a key role in diabetes mellitus–accelerated atherosclerosis clinical perspective. Circulation 2013;127:1888.
Zhang L-N, Wilson DW, da Cunha V et al, Endothelial NO synthase deficiency promotes smooth muscle progenitor cells in association with upregulation of stromal cell-derived factor-1α in a mouse model of carotid artery ligation. Arterioscler Thromb Vasc Biol 2006;26:765–772.
Lu H, Howatt DA, Balakrishnan A et al, Hypercholesterolemia induced by a PCSK9 gain-of-function mutation augments angiotensin II-induced abdominal aortic aneurysms in C57BL/6 mice-brief report. Arterioscler Thromb Vasc Biol 2016;36:1753–1757.
Shin IJ, Shon SM, Schellingerhout D et al, Characterization of partial ligation-induced carotid atherosclerosis model using dual-modality molecular imaging in ApoE knock-out mice. PLoS ONE 2013;8:e73451.
Schurmann C, Gremse F, Jo H et al, Micro-CT technique is well suited for documentation of remodeling processes in murine carotid arteries. PLoS ONE 2015;10:e0130374.
Nazari-Jahantigh M, Wei Y, Noels H et al, MicroRNA-155 promotes atherosclerosis by repressing Bcl6 in macrophages. J Clin Invest 2012;122:4190–4202.
Merino H, Parthasarathy S, Singla DK . Partial ligation-induced carotid artery occlusion induces leukocyte recruitment and lipid accumulation—a shear stress model of atherosclerosis. Mol Cell Biochem 2013;372:267–273.
Kanthi Y, Hyman MC, Liao H et al, Flow-dependent expression of ectonucleotide tri(di)phosphohydrolase-1 and suppression of atherosclerosis. J Clin Invest 2015;125:3027–3036.
Go YM, Kim CW, Walker DI et al, Disturbed flow induces systemic changes in metabolites in mouse plasma: a metabolomics study using ApoE(-)/(-) mice with partial carotid ligation. Am J Physiol Regul Integr Comp Physiol 2015;308:R62–R72.
Dunn J, Qiu H, Kim S et al, Flow-dependent epigenetic DNA methylation regulates endothelial gene expression and atherosclerosis. J Clin Invest 2014;124:3187–3199.
Alberts-Grill N, Rezvan A, Son DJ et al, Dynamic immune cell accumulation during flow-induced atherogenesis in mouse carotid artery: an expanded flow cytometry method. Arterioscler Thromb Vasc Biol 2012;32:623–632.
Sun X, Fu Y, Gu M et al, Activation of integrin α5 mediated by flow requires its translocation to membrane lipid rafts in vascular endothelial cells. Proc Natl Acad Sci USA 2016;113:769–774.
Heo K-S, Le N-T, Cushman HJ et al, Disturbed flow-activated p90RSK kinase accelerates atherosclerosis by inhibiting SENP2 function. J Clin Invest 2015;125:1299–1310.
Singh V, Rana M, Jain M et al, Curcuma oil attenuates accelerated atherosclerosis and macrophage foam-cell formation by modulating genes involved in plaque stability, lipid homeostasis and inflammation. Br J Nutr 2015;113:100–113.
Kumar S, Kim CW, Simmons RD et al, Role of flow-sensitive microRNAs in endothelial dysfunction and atherosclerosis: mechanosensitive athero-miRs. Arterioscler Thromb Vasc Biol 2014;34:2206–2216.
Kwak BR, Back M, Bochaton-Piallat ML et al, Biomechanical factors in atherosclerosis: mechanisms and clinical implications. Eur Heart J 2014;35:3020a–3020d.
Chlopicki S, Kozlovski VI, Lorkowska B et al, Compensation of endothelium-dependent responses in coronary circulation of eNOS-deficient mice. J Cardiovasc Pharmacol 2005;46:115–123.
Sun D, Liu H, Yan C et al, COX-2 contributes to the maintenance of flow-induced dilation in arterioles of eNOS-knockout mice. Am J Physiol Heart Circ Physiol 2006;291:H1429–H1435.
Gao G, Lu Y, Calcedo R et al, Biology of AAV serotype vectors in liver-directed gene transfer to nonhuman primates. Mol Ther 2006;13:77–87.
Acknowledgements
This work was supported by funding from National Institutes of Health Grants HL119798, HL113451, HL095070, and HL124879 to HJ. HJ is John and Jan Portman Professor. This project was supported in part by the Viral Vector Core of the Emory Neuroscience NINDS Core Facilities Grant, P30NS055077.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Author Contributions
SK, D-WK, and AR performed all the experiments. SK, D-WK, AR, and HJ analyzed the data. SK and HJ wrote the manuscript. HJ supervised the studies and secured funding. All authors reviewed the manuscript.
Supplementary Information accompanies the paper on the Laboratory Investigation website
The authors developed an accelerated mouse model of atherosclerosis using an adeno-associated virus containing mutant PCSK9 followed by partial carotid ligation surgery to induce hyperlipidemia, disturbed blood flow and atherosclerosis. This model enables rapid study of a gene-of-interest using transgenic or knockout mice without backcrossing them to ApoE or LDLR null background.
Supplementary information
Rights and permissions
About this article

Cite this article
Kumar, S., Kang, DW., Rezvan, A. et al. Accelerated atherosclerosis development in C57Bl6 mice by overexpressing AAV-mediated PCSK9 and partial carotid ligation. Lab Invest 97, 935–945 (2017). https://doi.org/10.1038/labinvest.2017.47
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2017.47
This article is cited by
-
PCSK9 stimulates Syk, PKCδ, and NF-κB, leading to atherosclerosis progression independently of LDL receptor
Nature Communications (2024)
-
Pla2g12b drives expansion of triglyceride-rich lipoproteins
Nature Communications (2024)
-
Myeloid CCN3 protects against aortic valve calcification
Cell Communication and Signaling (2023)
-
Flow-induced reprogramming of endothelial cells in atherosclerosis
Nature Reviews Cardiology (2023)
-
Endothelial discoidin domain receptor 1 senses flow to modulate YAP activation
Nature Communications (2023)
